The well-researched pro-dopamine regulator KB220 and variants result in increased functional connectivity in both animal and human brains, and prolonged neuroplasticity (brain cell repair) have been observed in rodents. Moreover, in addition to increased functional connectivity, recent studies show that KB220Z increases overall brain connectivity volume, enhances neuronal dopamine firing, and eliminates lucid dreams in humans over a prolonged period. An unprecedented number of clinical studies validating this patented nutrigenomic technology in re-balancing brain chemistry and optimizing dopamine sensitivity and function have been published. On another note, it is sad that unsuspecting consumers could be deceived and endangered by false promises of knock-off marketers with look-and-sound-alike products. Products containing ingredients having potential dangers (i.e., combinations of potent D2 agonists including L-Dopa and L-Threanine) threaten the credibility and reputation of validated, authentic, and ethical products. We encourage clinicians and neuroscientists to continue to embrace the concept of “dopamine homeostasis” and search for safe, effective, validated and authentic means to achieve a lifetime of recovery, instead of reverting to anti-dopaminergic agents doomed to fail in the war against the devastating drug epidemic, or promoting powerful D2 agonists that compromise needed balance.
D2 agonists, dopamine antagonists, dopamine homeostasis, drug epidemic, KB220, KB220Z
In 1908, President Theodore (Teddy) Roosevelt, worried that the national crisis of opiate addiction was weakening America and diminishing its greatness. Therefore, he appointed an Ohio doctor, Hamilton Wright, to be the nation’s first Opium Commissioner. In the decades after the Civil War, the United States developed a deadly narcotics habit. Suffering veterans were “hooked on morphine,” while genteel “society ladies” dosed up with laudanum—a tincture of alcohol and opium. The wonder drug was used widely as a cough suppressant, and proved very effective in treating diarrhea in children. In fact, in 1911, Wright told the NY Times —“Our prisons and our hospitals are full of victims of it; it has robbed ten thousand businessmen of moral sense and made them beasts who prey upon their fellows it has become one of the most fertile causes of unhappiness and sin in the United States”
Remarkably, more than a century later, America has relapsed [1]. The current opioid crisis is more lethal, with record numbers of fatal overdoses, public health professionals report. However, it is not the first time in U.S. history that the lax commercialization of legal opioids led to a national epidemic. To reiterate, faced with a late 19th-century dope scourge, federal law enforcement officials, doctors, and pharmacists eventually managed to contain the country’s first addiction epidemic [2].
The authors, who have numerous scientific publications on this issue, believe that FDA approved Medication Assisted Treatments (MATS), such as, the acute administration of buprenorphine, are helpful in inducing short-term dopamine release [3-5]. Chronic use, however, induces a significant reduction in dopamine release at the reward site of the brain, causing an unwanted, anti-reward state (see Figure 1) [4]. It is not yet definitively known what the exact biological consequences of chronic exposure to partial agonist or full antagonist, such as, in the form of buprenorphine or naltrexone, respectively, will be on brain function. It is important to consider that the current therapies require chronic receptor activity or blockades and are not natural means of restoring dopamine homeostasis.
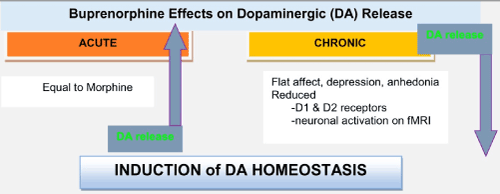
Figure 1. A graphical abstract showing short- and long-term administration of buprenorphine on dopamine release at the brain reward site (nucleus accumbens) [5].
For more than 50 years, our research has provided a significant dossier of peer-reviewed and published evidence in scientific journals showing that balancing dopamine dynamics in the brain reward circuitry is a far more desirable and useful strategy than blocking its normal physiologically required function [6]. Fundamentally, dogmatic protocols, such as methadone treatment, used routinely in addiction treatment are an attempt to medicate people with substance dependence back to health. To put it more simply, we are trying to force health rather than nourish the body’s ability to repair and rebalance via optimal gene expression [7]. This focus on medicating abstinence seems counterintuitive. Health can only be nourished, not forced; this is more easily said than done. In this multi-billion-dollar market, Big Pharma opted for simplicity, combating the global drug epidemic by blocking dopamine function with drugs like naltrexone (via mu receptor antagonism) or with, for example, Acamprosate, via antagonizing the NMDA–glutaminergic drive to release dopamine at the reward site (nucleus accumbens) [8].
Mark Gold and associates in their “dopamine depletion hypothesis” [9] suggested that the powerful dopamine two receptor (DRD2) agonist bromocriptine could be used for the treatment of cocaine addiction [10]. Fortunately, neuroscientists realized that chronic administration of powerful D2 agonists caused a severe reduction in the number dopamine receptors (down-regulation) [11]. The reason for this unwanted side effect is that bromocriptine or other powerful D2 agonists like L-Dopa overwhelm the neural pathways of the brain, especially the pleasure centers, and the biologically intelligent neurochemical adaptive mechanisms react to prevent too much dopamine function (hyper-dopaminergia) and, possibly, schizophrenic–like behaviors [12].
Blum and associates have been developing, by trial and error and genetic testing, a neuro adaptogen, KB220, and many subsequent improved variants, since 1968 [13]. To date, 37 published clinical studies have validated this nutrient technology based on gene mapping research. This patented technology is comprised of a list of ingredients intended to optimize gene expression and the synthesis, transport, reception, and disposal of neurotransmitters [14]. Optimization of gene expression for each neurotransmitter in the entire brain reward cascade, from serotonin in the brain stem to dopamine release in the nucleus accumbens/basal ganglia and cortical regions, achieving the functional ‘symphony of neurochemistry’ and the induction of “dopamine homeostasis” [15].
The first ever confirmed psychiatric genetic discovery by the Blum and Noble’s group, the association of the Dopamine D2 Receptor (DRD2) gene and severe alcoholism, was published in JAMA in 1990 [16]. The association in genetic studies of the DRD2 gene with many addictions, such as, alcohol, drugs, food, sex, nicotine, and other excessive reward seeking or self-medicating behaviors led to the idea of “Reward Deficiency Syndrome” (RDS), first coined by Kenneth Blum in 1995 [17]. Reward Deficiency Syndrome is now considered to be an established abnormal psychological syndrome listed in the SAGE Encyclopedia of Abnormal and Clinical Psychology (2017) [18] and refers to a deficiency of reward, paired with disrupted neurological dopamine function. This dysregulation of dopamine is the proposed cause of most (and, perhaps, all) all addictive, compulsive and impulsive behaviors.
To highlight the importance of the RDS concept, in 2013, B. William Downs and Kenneth Blum, published a paper entitled “Have We Hatched the Addiction Egg: Reward Deficiency Syndrome Solution System?" This paper was dedicated to all the people who have lost loved ones to substance abuse and "reward deficiency syndrome"- related tragedies. Why are we failing at reducing the incidence of RDS behaviors? Are we aiming at the wrong treatment targets? At that time, a paradigm shift was proposed; the "Reward Deficiency Solution System" and provided evidence for its adoption. The “Reward Deficiency Syndrome Solution System” and evidence was provided for the feasibility of its adoption [19,20]. While the RDSQ and GARS are in development and should be launched in 2018, the patented foundational ingredients of the KB220/Z/ZBR formulas have been studied in both animal (see Figure 2) [21] and human research (see Figure 3) [22]. After 50 years of study of brain reward systems, we now have fMRI evidence that KB220/Z/ZBR variants can enhance resting state, functional connectivity and brain connectivity volume (recruit neuronal firing in the reward center of the brain) [21] and balance the brain reward circuitry, especially, in abstinent heroin-dependent people.
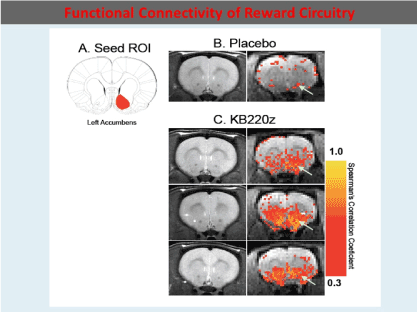
Figure 2. In rats KB220Z compared to Placebo seed Region of interest (ROI) os the left Accumbens
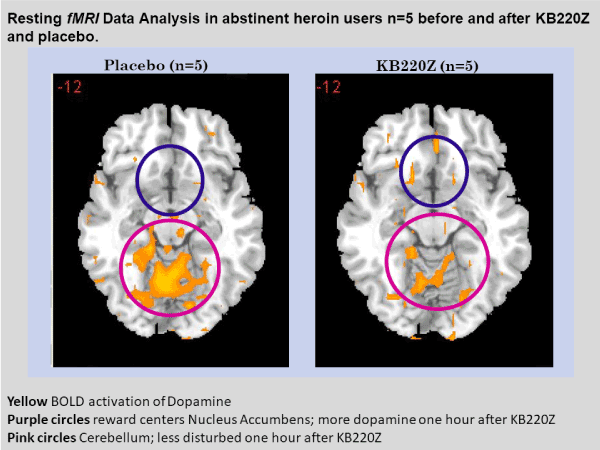
Unfortunately, a number of companies have decided to include L-Dopa as an ingredient in their nutraceutical to affect craving behavior. While the product contains some of the same ingredients found in KB220 variants, the inclusion of the drug L-Dopa, an aminoacid precursor of dopamine approved by FDA for Parkinsonism, with well documented side effects, is of concern. Of even greater concern is the potential of the tainted product to cause more harmful effects, common to products identified as “dietary supplements”. A possible harmful effect notwithstanding, this ingredient disrupts the neurochemical balance, especially of dopamine, and can induce unwanted hyperdopaminergia and dyskinesia, instead of much-needed balance [23]. It has the same effects as bromocriptine leading to dopamine D2 receptor down-regulation [24]. There is evidence that chronic administration of L-Dopa increases prefrontal cortex dopamine and serum corticosterone (a stress-related hormone) [25] and could enhance aberrant signaling in the D1 pathway [24] in denervation states, which arguably could be the case in the presence of chronic antagonists or reward deficiency. There is also evidence of a profound serotonin-dopamine imbalance, following L-Dopa treatment [26]. Some manufacturers, despite FDA restrictions, have produced plant-based L-Dopa in the form of Velvet Bean (Mucuna pruriens) and studies have linked this to both psychosis and homicidal behavior [27,28].
Over 100 million people in the United States carry the D2 receptor gene A1 allele [29], which is responsible for lower D2 receptor formation, is present in Parkinson’s disease and may be a precursor to the development of drug-induced dyskinesia, and potentially confer risk for Alzheimer’s disease [Blum et al., 2017]. Therefore, persons presenting for chemical dependency treatment should be warned about using any product containing L-Dopa. Moreover, low dopamine function can be problematic, specially, in carriers of the valine allele (replacement of methionine) that causes reduced dopamine function due to the high activity of synaptic dopamine break down [30]. This high activity could subsequently produce an unwanted potent neurotoxin metabolite from L-Dopa in the form of 3-O-methyldopa [31]. Another associated problem with L-Dopa administration is that it is known to cause a decrease in concentrations of S-Adenosyl methionine (SAM-e) [32] in cerebrospinal fluid with an increase in 3-methoxytyrosine, especially, in children. The small molecule, SAM-e, is involved in a process known as methyl donation, seen as an intermediate in one pathway to epigenetic cellular maintenance.
Known side effects of the chronic administration of L-Dopa for Parkinson’s patients include mania, dyskinesia (rigidity in extremities, face, mouth, and tongue), and abnormal involuntary movements (AIMs). Also, reported among the side effects are psychosis, auditory hallucinations, homicidality, hypersexuality, confusion, delusions, orthostatic hypotension, sleep disruption, age-related mental disturbances/cognitive decline, impaired gait, and kaliuresis (renal dysfunction with the induction of unwanted excretion of potassium) are side effects. Based on ignoring many studies, the use of L-Dopa is still considered Generally Recognized as Safe (GRAS), but the FDA has provided limitations on the over-the-counter use of L-Dopa and even the associated plant extracts [33]. Certain combinations that evoke significant caution include: threonine, a GRAS listed amino acid precursor, in combination with L-Dopa, are present in products that claim benefit for anti-cravings. L-Threanine increases neurotransmitter production, one of which is dopamine. L-Threanine (N-ethyl-L-glutamine) or theanine is an amino acid found in green teas. Historically, L-Threanine has been reported to be a relaxation-promoting agent. This has prompted some scientific research on its underlying pharmacology. Animal neurochemistry studies suggest that L-Threanine increases brain serotonin, dopamine, GABA levels and has micromolar affinities for AMPA, Kainate, and NMDA receptors [34]. Green tea has lots of threonines, although it can also be taken as a supplement. Along these lines, Acetyl-l-tyrosine (a potential supplement) is a production-ready form of tyrosine promoting brain dopamine synthesis. It is easy to understand that this combination of threonine and L-Dopa is unwanted, especially, in any nutraceutical supplement with the potential to impact the over-production of dopamine or even GABA, within the central nervous system and peripherally.
The well-researched, original pro-dopamine regulator, KB220, and variants (i.e. KB220Z/ZBR) utilizing recent technical advancements, show increased functional connectivity, in both animal and human neuroimaging studies. Prolonged neuroplasticity (neurogenesis) has been observed in rodents. Moreover, studies have been published showing that KB220Z increased function and brain connectivity volume, enhanced neuronal dopamine firing, and has eliminated lucid nightmares in humans over a prolonged period. An unprecedented number of clinical studies validating the success of a patented nutrigenomic technology to re-balance brain chemistry and optimize dopamine sensitivity and function have been published. The patented formula is the culmination of decades of meticulous trial and error tests on the effects of ingredients individually and on a plethora of combinations in animals and humans to determine the best behavioral benefits and outcomes. Subsequently, the formula outcomes were honed and verified by evaluating improvements in gene expression. Other ingredients have been included to expand the mechanistic reach to optimize gene expression for improved functionality of the neuro-endocrine-immune axis; optimizing the harmonious connectivity between different brain regions to achieve the symphony of neurochemistry. The improved effectiveness of the formulas has been further validated in clinical research including numerous brain scan studies published in peer-reviewed and cited scientific journals. Adding pro-hormones, plant-based hormonal analogs, or bio-identical hormonal substances to the exhaustively and meticulously researched and developed KB220Z/ZBR formulation will only disrupt the elaborate and sequential flow of genetic communication sequelae and the hamonious interconnectivity between neurotransmitters and their homeostatic, regulatory feedback controls in the Brain Reward Cascade. Due to these feedback regulatory controls, such additions result in homeostatic downregulation of neurotransmitter synthesis and reception to compensate for the artificial presence of such pseudo-neurotransmitter substances. It is a similar reactive mechanism, when prednisone is introduced to the system, the body stops making cortisone. The same mechanism applies to insulin, thyroid, and any neurotransmitter, especially, dopamine.
The KB220Z/ZBR nutrigenomic formulas (and prior variants) have been formulated to re-balance, harmonize, and optimize neurotransmitter functional relationships in the Brain Reward Cascade-not displace, replace, or disrupt those functional relationships. Moreover, products containing ingredients having potential dangers (i.e., combinations of potent D2 agonists, including L-Dopa and L-Threanine) threaten the credibility and reputation of validated, authentic, and ethical products and may cause unwanted harm to the unsuspecting subject. We encourage clinicians and neuroscientists to continue to embrace the concept of “dopamine homeostasis” and search for safe, effective, validated and authentic means to achieve a lifetime of recovery, instead of reverting to anti-dopaminergic agents doomed to fail in the war against this devastating drug epidemic.
2021 Copyright OAT. All rights reserv
All authors provided a substantial contribution to the conception, assisted with drafting the article, revising it critically, final approval of the version to be published, and agreement to act as a guarantor of the work.
The authors would like to thank Danielle Jean Kradin for formatting the references in the reference section. We appreciate Margaret A. Madigan for edits and figure configuration.
KB and MGL are recipients of grant 1R41MD012318-01. RDB is partially supported by the National Institutes of Health grants 1R01NS073884 and 1R21MH073624; and VA Merit Review Awards CX000479 and CX000780. The writing of this paper was supported in part by funds from the National Institutes of Health, NIAAA (RO1-AA07112 and K05-AA00219) and the Medical Research Service of the US Department of Veterans Affairs (MOB).
Kenneth Blum, Ph.D., is the holder of a number of U.S. and foreign patents issued and pending related to Nutrigenomics and Nutraceuticals. Dr. Blum is the Co-founder of Geneus Health LLC. Through Geneus Health, LLC, the Genetic Addiction Risk Score (GARS™) has licensed Dominion Diagnostics, LLC as a sales organization in the addiction space. Through his company Synaptamine, Inc., Dr. Blum licensed a number of retail outlets. He is a paid consultant of Dominion Diagnostics, LLC and The Shores Treatment & Recovery Center. Dr. Blum is a member of the scientific advisory board of Dominion Diagnostics, LLC, and is Chief Scientific Advisor of Dominion Diagnostics, LLC. He currently serves as Chairman of the Board and Chief Scientific officer, of Geneus Health, LLC. B. William Downs is CEO of Victory Nutrition International, Inc., and a licensee of the KB220Z/ZBR technology. There are no other author conflicts of interest.
- Al-Chalabi A, Hardiman O (2013) The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 9: 617-628. [Crossref]
- Renton AE, Chiò A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17: 17-23. [Crossref]
- Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, et al. (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18: 1226-1229. [Crossref]
- Nordin A, Akimoto C, Wuolikainen A, Alstermark H, Jonsson P, et al. (2015) Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum Mol Genet 24: 3133-3142. [Crossref]
- Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, et al. (2015) Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nature neuroscience 18: 1175-1182. [Crossref]
- Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, et al. (2015) C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol 14: 291-301. [Crossref]
- Satoh J, Yamamoto Y, Kitano S, Takitani M, Asahina N, et al. (2014) Molecular network analysis suggests a logical hypothesis for the pathological role of c9orf72 in amyotrophic lateral sclerosis/frontotemporal dementia. J Cent Nerv Syst Dis 6: 69-78. [Crossref]
- Xi Z, Zhang M, Bruni AC, Maletta RG, Colao R, et al. (2015) The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol 129: 715-727. [Crossref]
- Allen SP, Duffy LM, Shaw PJ, Grierson AJ (2015) Altered age-related changes in bioenergetic properties and mitochondrial morphology in fibroblasts from sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging 36: 2893-2903. [Crossref]
- Andrus PK, Fleck TJ, Gurney ME, Hall ED (1998) Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. Journal of neurochemistry 71: 2041-2048. [Crossref]
- Babu GN, Kumar A, Chandra R, Puri SK, Kalita J, et al. (2008) Elevated inflammatory markers in a group of amyotrophic lateral sclerosis patients from northern India. Neurochemical research 33: 1145-1149. [Crossref]
- Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, et al. (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137: 2329-2345. [Crossref]
- Chang Y, Kong Q, Shan X, Tian G, Ilieva H, et al. (2008) Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS One 3: e2849. [Crossref]
- Cozzolino M, Rossi S, Mirra A, Carrì MT (2015) Mitochondrial dynamism and the pathogenesis of Amyotrophic Lateral Sclerosis. Front Cell Neurosci 9: 31. [Crossref]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, et al. (2006) Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. Journal of neurochemistry 96: 1349-1361. [Crossref]
- Danzeisen R, Schwalenstoecker B, Gillardon F, Buerger E, Krzykalla V, et al. (2006) Targeted antioxidative and neuroprotective properties of the dopamine agonist pramipexole and its nondopaminergic enantiomer SND919CL2x [(+)2-amino-4,5,6,7-tetrahydro-6-Lpropylamino-benzathiazole dihydrochloride]. J Pharmacol Exp Ther 316: 189-199. [Crossref]
- Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, et al. (1997) Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. Journal of neurochemistry 69: 2064-2074. [Crossref]
- Finsterer J, Zarrouk-Mahjoub S (2016) Mitochondrial Disorders May Mimic Amyotrophic Lateral Sclerosis at Onset. Sultan Qaboos Univ Med J 16: e92-95. [Crossref]
- Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, et al. (2017) Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci Ther 23: 5-22. [Crossref]
- Hall ED, Andrus PK, Oostveen JA, Fleck TJ, Gurney ME (1998) Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. Journal of neuroscience research 53: 66-77. [Crossref]
- Higgins CM, Jung C, Xu Z (2003) ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci 4: 16. [Crossref]
- Jaiswal MK (2014) Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation. Mol Cell Ther 2: 26. [Crossref]
- Jiang Z, Wang W, Perry G, Zhu X, Wang X (2015) Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis. Transl Neurodegener 4: 14. [Crossref]
- Keeney PM, Bennett JP (2010) ALS spinal neurons show varied and reduced mtDNA gene copy numbers and increased mtDNA gene deletions. Molecular neurodegeneration 5: 21. [Crossref]
- Kong J, Xu Z (1998) Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. The Journal of neuroscience 18: 3241-3250. [Crossref]
- Ladd AC, Keeney PM, Govind MM, Bennett JP (2014) Mitochondrial oxidative phosphorylation transcriptome alterations in human amyotrophic lateral sclerosis spinal cord and blood. Neuromolecular Med 16: 714-726. [Crossref]
- Liu D, Wen J, Liu J, Li L (1999) The roles of free radicals in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB journal 13: 2318-2328. [Crossref]
- Malkki H (2015) Motor neuron disease: Brain transcriptome profiling reveals involvement of divergent pathways in C9orf72-associated and sporadic ALS. Nat Rev Neurol 11: 484. [Crossref]
- Martin LJ (2000) p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol Dis 7: 613-622. [Crossref]
- Martin LJ (2006) Mitochondriopathy in Parkinson disease and amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 65: 1103-1110. [Crossref]
- Martin LJ (2010) The mitochondrial permeability transition pore: a molecular target for amyotrophic lateral sclerosis therapy. Biochim Biophys Acta 1802: 186-197. [Crossref]
- Martin LJ (2011) Mitochondrial pathobiology in ALS. J Bioenerg Biomembr 43: 569-579. [Crossref]
- Sasaki S, Iwata M (2007) Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. Journal of neuropathology and experimental neurology 66: 10-16. [Crossref]
- Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, et al. (2016) The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med 22: 869-878. [Crossref]
- Zhao W, Beers DR, Appel SH (2013) Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. Journal of neuroimmune pharmacology 8: 888-899. [Crossref]
- Brohawn DG, O'Brien LC, Bennett JP (2016) RNAseq Analyses Identify Tumor Necrosis Factor-Mediated Inflammation as a Major Abnormality in ALS Spinal Cord. PLoS One 11: e0160520. [Crossref]
- Trias E, Ibarburu S, Barreto-Núñez R, Babdor J, et al. (2016) Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflammation 13: 177. [Crossref]
- Ladd AC, Brohawn DG, Thomas RR, Keeney PM, Berr SS, et al. (2017) RNA-seq analyses reveal that cervical spinal cords and anterior motor neurons from amyotrophic lateral sclerosis subjects show reduced expression of mitochondrial DNA-encoded respiratory genes, and rhTFAM may correct this respiratory deficiency. Brain Res 1667: 74-83. [Crossref]
- Huang da W, Sherman BT, Lempicki RA (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1-13. [Crossref]
- Huang da W, Sherman BT, Lempicki RA (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44-57. [Crossref]
- Marshansky V, Rubinstein JL, Gruber G (2014) Eukaryotic V-ATPase: novel structural findings and functional insights. Biochim Biophys Acta 1837: 857-879. [Crossref]
- Rawson S, Harrison MA, Muench SP (2016) Rotating with the brakes on and other unresolved features of the vacuolar ATPase. Biochem Soc Trans 44: 851-855. [Crossref]
- Walls KC, Ghosh AP, Franklin AV, Klocke BJ, Ballestas M, et al. (2010) Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J Biol Chem 285: 10497-10507. [Crossref]
- An T, Shi P, Duan W, Zhang S, Yuan P, et al. (2014) Oxidative stress and autophagic alteration in brainstem of SOD1-G93A mouse model of ALS. Mol Neurobiol 49: 1435-1448. [Crossref]
- Sasaki S (2011) Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 70: 349-359. [Crossref]
- Webster CP, Smith EF, Shaw PJ, De Vos KJ (2017) Protein Homeostasis in Amyotrophic Lateral Sclerosis: Therapeutic Opportunities? Front Mol Neurosci 10: 123. [Crossref]
- Otomo A, Pan L, Hadano S (2012) Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol Res Int 2012: 498428. [Crossref]
- Sioutas A, Vainikka LK, Kentson M, Dam-Larsen S, Wennerström U, et al. (2017) Oxidant-induced autophagy and ferritin degradation contribute to epithelial-mesenchymal transition through lysosomal iron. J Inflamm Res10: 29-39. [Crossref]
- Martin LJ (1999) Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol 58: 459-471. [Crossref]
- Seong E, Yuan L, Arikkath J (2015) Cadherins and catenins in dendrite and synapse morphogenesis. Cell Adh Migr 9: 202-213. [Crossref]
- Janzen C, Sen S, Lei MY, Gagliardi de Assumpcao M, Challis J, et al. (2017) The Role of Epithelial to Mesenchymal Transition in Human Amniotic Membrane Rupture. J Clin Endocrinol Metab 102: 1261-1269. [Crossref]
- Dalla Pozza E, Forciniti S, Palmieri M, Dando I (2017) Secreted molecules inducing epithelial-to-mesenchymal transition in cancer development. Semin Cell Dev Biol. [Crossref]
- Clark CR, Robinson JY, Sanchez NS, Townsend TA, Arrieta JA, et al. (2016) Common pathways regulate Type III TGFbeta receptor-dependent cell invasion in epicardial and endocardial cells. Cell Signal 28: 688-698. [Crossref]
- Cheng KY, Hao M (2017) Mammalian Target of Rapamycin (mTOR) Regulates Transforming Growth Factor-β1 (TGF-β1)-Induced Epithelial-Mesenchymal Transition via Decreased Pyruvate Kinase M2 (PKM2) Expression in Cervical Cancer Cells. Med Sci Monit 23: 2017-2028. [Crossref]
- Zhu FQ, Chen MJ, Zhu M, Zhao RS, Qiu W, et al. (2017) Curcumin Suppresses Epithelial-Mesenchymal Transition of Renal Tubular Epithelial Cells through the Inhibition of Akt/mTOR Pathway. Biol Pharm Bull 40: 17-24. [Crossref]
- Smith EF, Shaw PJ, De Vos KJ (2017) The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. [Crossref]