Paroxysmal Nocturnal Hemoglobinuria (PNH) is a rare disease caused by a somatic mutation in the PIG-A gene producing a deficiency of the complement regulatory proteins CD55 and CD59. This deficiency mainly affects the red blood cells causing hemolysis. Other clinical manifestations include thrombosis and bone marrow failure. Nearly 20% of classic PNH patients develops pancytopenia within 10 years after diagnosis, condition associated with worse outcome.
Eculizumab is a monoclonal antibody that inhibits the complement cascade by binding to complement protein C5 leading to a rapid and clinically significant reduction of intravascular hemolysis and other PNH complications, reducing the frequency of severe and life-threatening morbidities, such as thrombosis; and, consequently, improving patient´s survival. Eculizumab is considered the drug of choice for long-term treatment of classic PNH patients. Hematopoietic stem cell transplantation (HSCT) is a therapeutic option for PNH patients who developed severe aplasia. However, information regarding HSCT for Eculizumab treated patients is scarce.
In this manuscript, we report a rare case of a patient with PNH associated with aplastic anemia that was treated with Eculizumab and HSCT, and we review the indication of treatment for these patients, optimal duration of Eculizumab therapy after HSCT and safety of this treatment.
paroxysmal nocturnal hemoglobinuria, aplastic anemia, eculizumab, hematopoietic stem cell transplantation
A 26-yr-old male patient was initially referred to the hospital with fever, malaise and mucosal bleeding associated with profound pancytopenia in June 1993. A severe aplastic anemia (AA) was diagnosed in 1993. Due to the fact that he did not have an HLA identical sibling, he received immunosuppressive treatment (using anti-thymocyte globulin (ATG) in combination with cyclosporine A (CsA) and steroids) and a partial remission was observed.
In June 2005, after an intravascular hemolysis episode (reticulocytosis, elevated LDH and hemosideruria) and a flow cytometry study of GPI-proteins, the patient was diagnosed of PNH with high hemolytic activity. In 2008, patient treatment was started upon Eculizumab availability following the standard schedule (600 mg weekly for 4 weeks, followed by 900 mg every other week) and resulting in a remarkable reduction of the hemolytic activity and normalization of hemoglobin (Hb) levels within a few months. Hematologic parameters remained stable until January 2014, when a severe neutropenia and thrombocytopenia was observed. Bone marrow biopsy revealed severe AA (with no chromosomal abnormalities), with absolute neutrophil count 0.5x109/L; Hb 5.0 g/L, and platelets 20x109/L. The patient received a second course of immunosuppressive therapy (IST) in July 2014, showing partial remission. Because of the treatment, the patient suffered several infections including Influenza A, Pseudomona sepsis and cholecystitis by Serratia and Enterococcus.
The 9th of June of 2015 (day 0) he received a hematopoietic stem cell transplant from an ABO-matched and 9/10-allele HLA-matched (mismatch in HLA-C) CMV-negative female donor. At this moment, the fluorimetric study showed a percentage of RBC type III of 75.9%, neutrophils CD16-FLAER- of 99.2%, and monocytes CD14-FLAER- of 97.3% (Figure 1). The unmanipulated bone marrow contained only 0.97×106 CD34+ cells/kg body weight. The conditioning regimen included fludarabine (30 mg/m2 on days –5 through –3), cyclophosphamide (30 mg/kg on days -5 to -2) and ATG (2.5 mg/kg on days –3 through –1). Graft-versus-Host-Disease (GVHD) prophylaxis included CsA i.v. (target level 200-300 ng/mL) and methotrexate (15 mg/m2 day 1, 10 mg/m2 days +3, +6, +11).
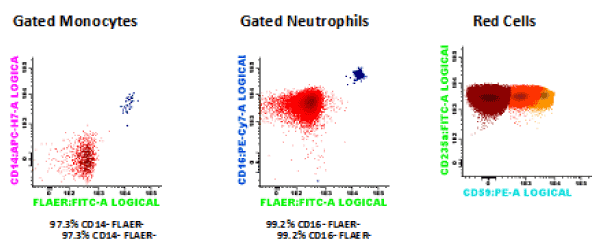
Figure 1. Flow cytometry at the moment of the transplantation: RBC type III 75.9%, neutrophils CD16- FLAER- 99.2%, monocytes CD14-FLAER- 97.3%.
Eculizumab treatment was maintained due to the presence of a significant PNH clone population and the high risk of breakthrough hemolysis and thromboembolic events until engraftment (last dose 8th of July 2015). Complement activity assay CH50 remained undetectable during Eculizumab administration and hemolysis was not observed throughout the course of the transplantation.
During the first two months after HSTC, the pancytopenia gradually decreased and no hemolysis was observed (Figure 2). A trephine biopsy showed hyper-cellularity and the proportion of PNH clone decreased (RBC type III of 6.43%, neutrophils CD16-FLAER- of 0.43% and monocytes CD14-FLAER- of 21%). Further, the complete donor chimerism was achieved. The patient did not present acute or chronic GVHD and CsA was tapered starting on day 29 of February 2016 and discontinued two months later.
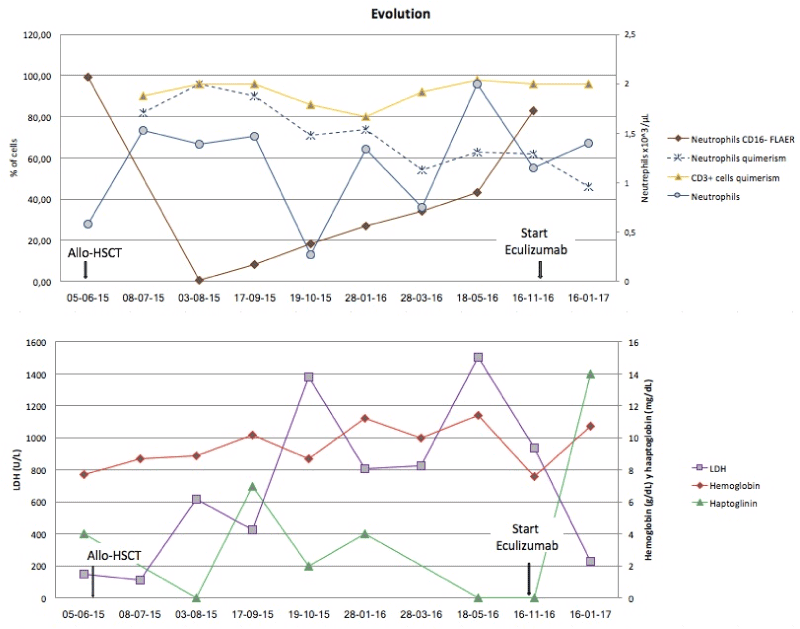
Figure 2. The kinetics of PNH neutrophils, donor chimerism in neutrophils and T cells and the levels of LDH, hemoglobin and haptoglobin.
Six months after the transplantation, the patient presented a long lasting and progressive pancytopenia associated with an increased PNH clone and a slowly loss of chimerism. 17 months after HSTC, the patient showed again a severe asthenia, esophageal spasm with Hb 7.8 gr/dL and LDH 2343 U/L. The flow cytometry study of GPI-AP expression revealed a percentage of RBC type III of 59.3%, neutrophils CD16-FLAER- of 83% and monocytes CD14-FLAER- of 95%. The patient was vaccinated against Neisseria meningitides and treated with Eculizumab in November 2016. His fatigue dramatically improved and the level of Hb gradually increased. Eculizumab was well tolerated resulting in a remarkable reduction of the hemolytic activity and a rapid reduction of LDH levels, acquiring transfusion independence since the beginning of the treatment [1,2].
PNH is a very rare clonal disease of hematopoietic stem cell characterized by intravascular hemolysis, thrombosis and bone marrow failure with an incidence of 1.3 per million population per year. AA is also a rare disease that has an incidence of 1-2 per million population per year. Nevertheless, a significant overlap between the two conditions exists: at least 50% of AA patients have a PNH clone and 40% of PNH patients develop some grade of bone marrow failure.
Treatment options for PNH included Eculizumab and HSCT. Eculizumab is effective in reducing intravascular hemolysis and risk of thrombosis, but it does not improve bone marrow failure in PNH. Allogeneic HSCT is also able to eradicate the PNH clone, nevertheless it is associated with a significant morbidity and mortality, not all patients have an HLA-matched donor available and acute and chronic GVHD occurs in a third of the patients. Survival analysis of 26 patients who underwent HSCT in Italy from 1988 to 2006 revealed a 10-yr disease free survival of 57% [3]. Before Eculizumab, the presence of each clinical hallmark of the disease (hemolytic anemia, thrombosis and marrow failure) may qualified patients for transplantation, especially the younger ones. Nowadays, the indication of HSCT has changed, but they are still a matter of intense discussion.
In a large retrospective study of 211 patients transplanted for PNH in 83 EBMT centers from 1978 to 2007: the 5 yr-OS probability was 86% ± 5 in patients with recurrent hemolytic anemia, 54% ± 7 in patients with thromboembolism, and 69% ± 5 in patients with AA [4]. The excellent overall survival [5] and improvement of the quality of live [6] in PNH patients with recurrent hemolysis have favored the use of Eculizumab. In the EBMT study, 30% of transplanted patients developed chronic GVHD with a significant alteration of the quality of life [4]. Consequently, Eculizumab is now considered as the therapy of choice for treating transfusion-dependent and symptomatic patients that show intravascular hemolysis and related manifestations.
Thromboembolism in PNH was associated with worse outcome of transplanted patients and transplantation is probably not a suitable option for these patients due to an unacceptably high toxicity. Thus, the presence of thrombosis was associated with a high mortality rate in other retrospective study [7]. Given its efficacy in preventing thromboembolism, Eculizumab may also be considered as suitable option for PNH patients with recurrent thrombosis, regardless of the symptoms, hemolysis and, even, the PNH clone size.
Nowadays, there is a paucity of available data to guide the treatment of AA and PNH. It is currently recommended to treat patients with PNH and AA based on the guidelines for AA. IST therapy remains as the treatment of choice for patients who lack an HLA identical sibling donor. However, 30-40% of the patients will relapse or will be refractory to IST and they will be considered for transplantation using an alternative donor. Consequently, HSCT should be restricted to patients with an underlying bone marrow failure. In AA patients with a small PNH clone, with thrombosis and without hemolysis, the standard treatment should be similar to that of patients with AA instead of patients with PNH [8].
Some concerns regarding the management of PNH in patients with severe AA, such as the optimal duration of Eculizumab therapy after HSCT and the safety of this treatment, exist. Nevertheless, Eculizumab may have a positive effect in the amelioration of inflammation and the hemostatic activation and might be effective for preventing thrombotic events as microangiopathy during HSCT. Contrarily, discontinuation of Eculizumab treatment may result in recurrence of hemolysis and increased bilirubin levels and it may complicate the management of early course HSCT and the immunosuppressive therapy. Even so, some authors recommend the discontinuation of Eculizumab treatment before the initiation of ATG to avoid a diminished efficacy of ATG due to complement mediated cytotoxicity (CDC). Nonetheless, the effects of terminal complement inhibition on engraftment or GVHD remain unknown. Loss of chimerism in our patient was probably related with number of CD34 cells infused. GVHD remains a major cause of morbidity and mortality after HSCT and without advantages in no malignant disorders such as PNH and AA. The use of ATG is associated with reduced risk of acute and chronic GVHD in AA [9].
The use of IST with ATG in combination with Eculizumab in PNH/AA patients has been limited to a few case reports and no reduced ATG efficacy or adverse event was observed [10-12]. There are few cases reporting PNH/AA patients receiving Eculizumab therapy before HSCT. In one patient, Eculizumab was stopped 1 month before transplantation to avoid reducing the efficacy of CDC by ATG used in the conditioning regimen, but the CH50 level was still low on the day of starting the conditioning regimen. The effect of ATG was probably mediated for other mechanisms different from CDC [13], suggesting a major role for antibody-dependent cellular cytotoxicity or even programmed cell death [14]. In other case report, Eculizumab was discontinued 2 weeks prior to the initiation of the conditioning regimen and this was associated with a highly lymphoablative conditioning regimen for achieving rapid and complete donor chimerism and a rapid decrease of the proportion of PNH erythrocytes [15]. Another patient received Eculizumab prior to and immediately following allogeneic HSCT (1-day post transplantation because a hemolytic episode occurred) without an adverse effect on engraftment or clinical outcome [16]. The largest cohort of patients receiving concurrent therapy from both AA and PNH was reported from UK PNH National Service. The conclusions of this study were that the response rates for IST in patients with Eculizumab were similar compared with age matched controls and the presence PNH clone should not influence AA treatment decisions [17]. A recommendation of this study was that Eculizumab may be stopped in patients undergoing bone marrow transplant once the PNH white cell clone has disappeared, but these recommendations are based in only 2 transplanted patients [18].
The decision regarding the best time of Eculizumab discontinuation after HSCT is a dilemma because it could either have an impact on engraftment or result in development of severe infections. Eculizumab blocks terminal complement activation and increases the susceptibility to infections by blocking lysis of bacteria, especially with encapsulated bacteria. Infection is known to be one main cause of mortality in patients that continue with Eculizumab therapy after transplantation since it may result in a 2-fold increase of infection susceptibility.
An involvement of complement dysregulation in developing transplant-associated thrombotic microangiopathy (TA-TMA) was recently suggested [19]. While pathogenesis of TA-TMA following HSCT is poorly understood, accumulating evidence suggests a role for complement activation on endothelial cells. A prospective study compared children and young patients with high-risk TMA following HSCT who were treated with Eculizumab with a cohort of historical controls who did not receive Eculizumab [20]. 61% of Eculizumab-treated patients achieved resolution of TMA and 1yr-OS after the transplantation was clearly superior in the Eculizumab-treated group (61% vs 9%). All patients received prophylaxis against Neisseria meningitides during Eculizumab therapy and the infection rates were similar in treated and untreated patients. These data indicate that terminal complement blockage without vaccination in the early post-transplant period may be performed as long as the appropriate prophylaxis is used until complement function is restored.
The manufacturer recommends the immunization of the patients with the meningococcal vaccine at least 2 weeks before starting the therapy. However, this may not be feasible in HSCT recipients because they do not have adequate immunity. For these patients, anti-meningococcal antibiotics (ciprofloxacin or penicillin VK) may be considered as an alternative prophylactic approach.
In the next years, it is expected an increase of life expectancy of PNH patients receiving Eculizumab treatment increasing the possibility of development of bone marrow failure which will probably increase the treatment of concurrent PNH and AA patients. Given the rarity of the disease, worldwide cooperative studies are desirable to compare HSCT, or more conservative treatments, in association with Eculizumab, as well as to investigate specific issues related to the transplant procedure in PNH patients.
In our case, we observed that Eculizumab administration did not negatively affect the development of GVHD or infection after HSCT and we show that HSCT may not be curative in all patients. Thus, Eculizumab should be considered as a safe alternative for improving survival of these patients.
2021 Copyright OAT. All rights reserv
This work was supported by the Spanish grant of Instituto de Salud Carlos III (PI16/01485) and FEDER European Union. SLH hold a Severo Ochoa Grant (BP14-150). The authors have no conflicts of interest to disclose.
- de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, et al. (2008) Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood 112: 3099-106. [Crossref]
- Hillmen P, Muus P, Röth A, Elebute MO, Risitano AM, et al. (2013) Long‐term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol 162: 62-73. [Crossref]
- Santarone S, Bacigalupo A, Risitano AM, Tagliaferri E, Di Bartolomeo E, et al. (2010) Hematopoietic stem cell transplantation for paroxysmal nocturnal hemoglobinuria: long-term results of a retrospective study on behalf of the Gruppo Italiano Trapianto Midollo Osseo (GITMO). Haematologica 95: 983-8. [Crossref]
- de Latour RP, Schrezenmeier H, Bacigalupo A, Blaise D, de Souza CA, et al. (2012) Allogeneic stem cell transplantation in paroxysmal nocturnal hemoglobinuria. Haematologica 97: 1666-73.
- Kelly RJ, Hill A, Arnold LM, Brooksbank GL, Richards SJ, et al. (2011) Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood 117: 6786-92. [Crossref]
- Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, et al. (2006) The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med 355: 1233-43. [Crossref]
- Kamranzadeh Fumani H, Zokaasadi M, Kasaeian A, Alimoghaddam K, Mousavi SA, et al. Allogeneic hematopoietic stem cell transplantation for paroxysmal nocturnal hemoglobinuria: a retrospective single‐center study. Hematol Oncol 1: 1-41. [Crossref]
- Peffault de Latour R (2016) Transplantation for bone marrow failure: current issues. Hematology Am Soc Hematol Educ Program 90-98. [Crossref]
- Bacigalupo A, Socié G, Hamladji RM, Aljurf M, Maschan A, et al. (2015) Current outcome of HLA identical sibling versus unrelated donor transplants in severe aplastic anemia: an EBMT analysis. Haematologica 100: 696-702. [Crossref]
- Alashkar F, Dührsen U, Röth A (2016) Horse anti‐thymocyte globulin and eculizumab as concomitant therapeutic approach in an aplastic paroxysmal nocturnal hemoglobinuria patient: go or no‐go?. Eur J Haematol 97: 403-5. [Crossref]
- Asano J, Ueda R, Tanaka Y, Shinzato I, Takafuta T (2013) Effects of immunosuppressive therapy in a patient with aplastic anemia-paroxysmal nocturnal hemoglobinuria (AA-PNH) syndrome during ongoing eculizumab treatment. Intern Med 53: 125-8. [Crossref]
- Marotta S, Giagnuolo G, Basile S, Pagliuca S, Grimaldi F, et al. (2014) Excellent outcome of concomitant intensive immunosuppression and eculizumab in aplastic anemia/paroxysmal nocturnal hemoglobinuria syndrome. Journal of Hematology & Thromboembolic Diseases 2: 1.
- Oshiro H, Goi K, Akahane K, Inukai T, Sugita K (2015) Effective eculizumab therapy followed by BMT in a boy with paroxysmal nocturnal hemoglobinuria. Pediatr Int 57: e27-9. [Crossref]
- Goh BK, Chedid MF, Gloor JM, Raghavaiah S, Stegall MD (2012) The impact of terminal complement blockade on the efficacy of induction with polyclonal rabbit antithymocyte globulin in living donor renal allografts. Transpl Immunol 27: 95-100. [Crossref]
- Taniguchi K, Okada M, Yoshihara S, Sawada A, Tokugawa T, et al. (2011) Strategy for bone marrow transplantation in eculizumab-treated paroxysmal nocturnal hemoglobinuria. Int J Hematol 94: 403-407. [Crossref]
- Goker H, Uz B, Buyukasik Y, Aksu S, Haznedaroglu I, et al. (2011) Eculizumab before and after allogeneic hematopoietic stem cell transplantation in a patient with paroxysmal nocturnal hemoglobinuria. Turk J Hematol 28: 223-7. [Crossref]
- Griffin M, Kulasekararaj A, Gandhi S, Hillmen P, Munir T, et al. (2016) Concurrent Treatment of Aplastic Anaemia (AA) with Immunosuppressive Therapy and Paroxysmal Nocturnal Hemoglobinuria (PNH) with Eculizumab: A UK Experience. Blood 128: 2683.
- Griffin M, Hillmen P, Munir T, Richards S, Arnold L, et al. (2016) Concurrent treatment of aplastic anemia (AA) with immunosupressive therapy and paroxysmal nocturnal hemoglobinuria (PNH) with eculizumab. Haematologica 101: 612.
- Jodele S, Zhang K, Zou F, Laskin B, Dandoy CE, et al. (2016) The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood 127: 989-96. [Crossref]
- Jodele S, Fukuda T, Mizuno K, Vinks AA, Laskin BL, et al. (2016) Variable eculizumab clearance requires pharmacodynamic monitoring to optimize therapy for thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 22: 307-15. [Crossref]