Endomyocardial diseases are a rare and poorly understood aetiology of cardiomyopathy (CM). Common endomyocardial diseases capable of causing CM are endomyocardial fibrosis (EMF), hypereosinophilic syndrome (HES) with cardiac involvement and endocardial fibroelastosis (EFE). The key pathogenic mechanisms of HES and EMF are sustained eosinophilia while EFE is cardiac anomalies, infections or genetics causing fibroelastic thickening of the endocardium and systolic dysfunction. However, the recent classifications by the American Heart Association and the European Society of Cardiology describe CM due to endomyocardial diseases within the restrictive CM phenotype yet they might be two clinically distinct entities. Endomyocardial diseases as a cause of CM also lack sufficient evidence to develop a clear understanding of their natural course, diagnosis and management. Available evidence rely largely on earlier studies that have not incorporated progressive changes in non-invasive cardiac imaging that have occurred over the decades. Improved understanding of the disease and diagnosis is essential since early diagnosis and treatment targeted at the cause is essential to improve efficacy and survival. This paper therefore reviews published data on CMs due to endomyocardial diseases with a focus on epidemiology, aetiology, pathological features, clinical presentation, diagnosis and management. Improved knowledge on aetiologies of CMs such as endomyocardial diseases is important to develop targeted treatment and improve both patient and clinical outcomes.
endomyocardial fibrosis, eosinophilic cardiomyopathy, endocardial fibroelastosis, hypereosinophilic heart disease
Cardiomyopathies (CM), as a non-coronary cause of heart failure (HF), was established in the 18th Century, and subsequently classified into four types: dilated (DCM), hypertrophic (HCM), restrictive (RCM) and (more recently) arrhythmogenic cardiomyopathy [1-3]. Towards the end of the 20th Century, the availability of sensitive cardiac imaging modalities and advancements in genetic testing further allowed the sub-division of the four major CM categories by aetiology and pathogenesis [4-6]. In terms of aetiology-based classification, recent review articles on CMs have described endomyocardial diseases as an aetiology of restrictive or infiltrative CM [7,8]. The lack of consistency in the classification of CM due to endomyocardial diseases undermines comparative studies and subsequently a holistic understanding of its clinical course, pathophysiology and clinical management. In addition, due to the rarity of the CM due to endomyocardial diseases, there is a clear lack of evidence on diagnosis and management yet the disease has an ominous prognosis if not detected early and prompt treatment initiated. This present systematic review and meta-analysis aggregates published evidence on endomyocardial diseases as a cause of CMs with a particular focus on pathophysiology, diagnosis and clinical management. This review also identifies grey areas that might benefit from further research to improve current understanding of endomyocardial diseases CMs.
Endomyocardial diseases refer to a group of diseases that affect the endocardium and/or myocardium leading to myocardial injury that may range from a fully recoverable syndrome to one that leads to chronic myocardial remodelling and restrictive cardiomyopathy (RCM) [7,8]. Generally, endomyocardial diseases associated with CMs define two major variants of RCM (endomyocardial fibrosis and hypereosinophilic heart disease), which exhibit overlapping pathological features and prominent eosinophilic involvement yet they are likely two distinct clinical entities. A third closely related disease affecting the endocardium and capable of causing CM is endocardial fibroelastosis (EFE), whose cause is distinctively different from endomyocardial fibrosis and hypereosinophilic heart disease but with almost indistinguishable key pathological features [8].
Endomyocardial fibrosis
Epidemiology : Endomyocardial fibrosis (EMF; formerly Davies disease) is a rare form of myocardial disease usually seen in the elderly or is endemic and affects mainly children and adolescents usually characterized by the deposition of fibrous tissues in the endomyocardium leading to restrictive physiology [9-11]. Since the seminal description by Davies in Uganda in 1948, the disease has a high frequency in resource-constrained tropical regions of Africa, Latin America and Asia making it the leading cause of RCM in countries in these endemic regions [12-14]. In sub-Saharan Africa, EMF is predominant in Uganda, along the low-lying coastal belt of Mozambique and in some parts of West Africa, with sporadic cases reported in Congo and Malawi [9]. In these endemic regions of Africa, EMF if the main cause of HF accounting for up to 20% of all cases [15,16]. A recent screening study in Mozambique reported about 20% prevalence in a random sample of 1,063 subjects of all age groups who had echocardiographic evidence of the EMF [17].
In addition to the tropical regions of Africa, India has a high burden of EMF along the coastal area and rain forest of Kerala State while China has a high prevalence in the province of Guangxi [18-21]. In Latin America, reports of EMF have been made in Brazil and Colombia [22,23]. A decline in India and some regions of Nigeria is in contrast with persistent high trends in other areas suggesting a potential influence of socioeconomic and environmental factors on the disease [9]. Data on sex distribution are mixed. In Uganda, women of childbearing age have a two-fold higher prevalence compared to men, male preponderance in Mozambique and Nigeria while other studies have not observed a specific sex difference in adults [16,17,24-27]. Long-term outcome from medical treatment in advanced stages is very poor with 75% mortality at two years [28]. Overall, EMF accounts for about 20% of hospitalization for HF in Nigeria, Equatorial Guinea and Uganda, where the disease is the leading cause of paediatric admission for acquired heart disease only second to rheumatic heart disease [29-32].
Aetiopathogenesis: The aetiopathogenesis of EMF remain unclear, warranting more systematic research and the use of contemporary technologies to test older classical hypothesis whose findings have varied across studies. The proposed causes or co-factors for the development of EMF include poverty, malnutrition, parasitic infestation, genetics and cluster ethnicity while proposed theories for the pathogenesis of EMF are eosinophilia, infectious disease and autoimmunity [9,33-37]. Similarities in cardiac lesions with hypereosinophilic syndrome (HES) and the prevalence of parasites in the EMF endemic countries suggest eosinophilia toxicity and infections as potential primary aetiologic agents [9,19,20,35-39]. However, variable association with eosinophilia and the frequent absence of eosinophils on endomyocardial biopsy, even in the early stages of EMF, may argue against the involvement of eosinophilia [10,39-44].
A recent hypothesis suggests an infective trigger in genetically susceptible individuals [44]. Excessive immune response may be the link between certain parasitic infections and EMF. Already, there are reports of increased circulating levels of antibodies IgE and manifestations of hyper-immune malaria-related splenomegaly among Rwandan immigrants with EMF to support the infective trigger hypothesis [45-47]. Immunological investigations also reveal the presence of autoantibodies IgG and IgM directed against myocardial proteins [48,49]. However, inconsistent geographical matching with important parasitic infections such as schistosomiasis and filariasis particularly in Southeast Asia, and the lack of significant differences in parasitic loads between EMF patients compared with controls provide an argument against a straightforward parasitic-related immunological pathogenic mechanisms [16,50-53]. Molecular mimicry similar to that observed in patients with rheumatic heart disease has also been suggested as a possible pathogenic mechanism but lacks compelling evidence [54-56].
The involvement of environmental factors, dietary factors and toxins have also been suggested as possible pathogenic agents of EMF. Several environmental (toxic) factors such as magnesium deficiency, cerium toxicity, cyanogenic glycosides, high vitamin D serotonin toxicity and certain herbal preparations have been proposed to be involved in the pathogenesis of EMF but they lack compelling supportive evidence [9]. In certain genetically predisposed individuals, poor diet may lead to dysfunction in the regulatory and functional mechanisms of eosinophilic leukaemia resulting in necrosis, thrombosis and fibrosis, supported by evidence that long-standing dietary imbalance with low-protein intake has been observed in EMF patients [57].
A convincing diet-related hypothesis is that associated with cassava consumption, which is a staple food in most EMF endemic areas of Africa and India. Cassava contains linamarin, a toxic cyanogenic glycoside, which can liberate hydrogen cyanide in the gut during digestion. In foods that have not been properly processed or cooked, toxic levels of glycoside may persist in the tuber during consumption. The human body detoxifies cyanide by converting it to thiocyanate via the sulphur containing enzyme rhodanase. A low-protein diet deficit in sulphur containing amino acids may decrease the detoxification capacity thereby increasing vulnerability to the toxic effects of cyanide, which may be compounded by excessive cassava consumption as the sole source of dietary energy and protein [58]. Sub-lethal doses of cyanide levels may result in tissue hypoxia and lipid peroxidation, which alters myocardial cell biology as observed in neuronal toxicity [59].
Consistent with the cassava pathogenic hypothesis, animal models have associated cassava intake with the development of intracellular vacuoles, endocardial thickening and interstitial fibrosis that is independent of eosinophilic toxicity or parasitic infestation [9]. Improved socioeconomic status accompanied by a significant drop in cassava intake have been associated with a marked decline in cases of EMF in Kerala, India supporting the role of cassava in the pathogenesis of EMF. However, a mismatch between the distribution of EMF and malnutrition in Africa, and incidences of EMF among subjects from non-tropical areas who spent a short period in endemic areas is inconsistent with the cassava hypothesis [43,60-62]. Finally, a high prevalence of right-sided HF suggest the involvement of toxic factors removed by the lung in EMF patients [9].
Natural course: The hallmark clinical feature of EMF is patchy fibrosis of the endocardial cardiac surface, which may lead to decreased compliance and ultimately restrictive physiology with a more general involvement of the endomyocardial surface [33]. Endocardial fibrosis largely involves the apices and inflow tracts of the RV and/or LV and may affect atrioventricular (AV) mainly through tethering the papillary muscles, leading to tricuspid and/or mitral regurgitation [63]. Olsen proposed three pathomorphological phases of the EMF in his patients from Uganda. The initial (or acute necrotic) phase involves eosinophilic infiltration of the myocardium with necrosis of the sub-endocardium with a pathological picture consistent with acute myocarditis (MC) characterized by febrile illness and in severe cases, HF and shock. This initial phase occurs in the first five weeks of illness [64]. Patients who survive this acute initial phase progress into the second stage, typically presenting after ten months characterized by thrombus formation over the initial lesions with a decrease in inflammatory activity. Ultimately, after several years of disease activity, the final fibrotic phase manifests, when the collagenous fibrosis replaces the endocardium [33].
The Olsen three-phase pathomorphologic classification of EMF does not apply uniformly to all EMF patients and other investigators have not provided consistent support. Most of the patients remain asymptomatic for long periods and often present with the chronic burnt-out phase with isolated endocardial involvement and intracardiac thrombi [65]. Upon clinical diagnosis, the onset of complication such as atrial fibrillation (AF), thromboembolism and progressive AV valve regurgitation abbreviates the natural history [66]. Myocardial fibrosis consists of collagen deposition and fibroblast proliferation, which explains most of the symptoms in EMF patients [33]. Fibrosis increased stiffness of the heart leading to restrictive physiology. Ventricular stiffness along with AV valvular regurgitation leads to atrial enlargement associated with atrial arrhythmias such as AF. Fibrosis also causes decreased conduction velocity, impaired activation patterns and may provide substrate for wave breaks and re-entry [67]. AF affects more than 30% of EMF patients followed by other rhythm and conduction aberrations such as junctional rhythm, heart blocks and AV conduction delays [68].
Clinical presentation: Clinical presentation of EMF includes an insidious onset although may be heralded by acute febrile illness. Symptoms at presentation may relate to specific cardiac chambers and/or valves where the disease is most extensive, the duration of the disease and the presence of signs of activity [69]. Pulmonary congestion is a sign of left-sided involvement while predominant right-sided disease may mimic RCN or constrictive pericarditis. AV valve regurgitation is common. Often, EMF is relentless and progressive although the time course of decline may vary considerably. Cachexia, malnutrition and hypoalbuminemia are characteristic of advanced disease. Common modes of death are progressive HF, arrhythmias, infection, infarction, sudden cardiac death and complications of surgery [70]. Clinical presentation may differ based on RV, LV or bi-ventricular involvement [33].
In predominant or pure RV involvement, fibrous tissues cover the RV apex, and these tissues may extend to the tricuspid valve with ensuing tricuspid regurgitation. There is manifestations of chronic systemic venous hypertension and exophthalmos, elevated jugular venous pressure, prominent v wave with rapid y descent and a right-sided S3 gallop. Prominent hepatomegaly with pulsatile liver, ascites, splenomegaly and peripheral oedema but pulmonary congestion is absent because the left side of the heart is not involved explaining the normal pulmonary artery and pulmonary capillary wedge pressure in the affected patients. A large pericardial effusion is often present but with pleura spared, and (may be) dilated right atrium (RA) [71]. Distinctive features not explained solely by low cardiac output and retrograde congestion include central cyanosis, giant ascites in the absence of pedal oedema, hyper-pigmentation of lips and gum, proptosis and parotid swelling [72]. ECG abnormalities are consistent with RA enlargement such as supraventricular arrhythmias, AF, low QRS voltage, AV blocks, RBBB or LBBB or non-specific ST-T wave changes. Chest radiography reveals RA prominence, pericardial effusion and calcification of RV and (less frequently) LV wall. On echocardiography, RV thickening with obliterated apex, dilated atrium, hyperechoic endocardial surfaces and abnormal septal motion in patients with tricuspid regurgitation but with spared outflow tracts. Doppler echocardiography reveals typical restrictive filling pattern (increased E/A ratio, and decreased deceleration time and isovolumic relaxation time [IVRT]) [33].
EMF with predominant LV involvement manifests with fibrotic involvement of the ventricular apex, the chordae tendineae or posterior mitral valve leaflet producing mitral regurgitation. The associated murmur may be late systolic suggesting papillary muscle dysfunction murmur or pansystolic. Prominent pulmonary hypertension and S3 protodiastolic gallop is frequently present [73]. ECG abnormalities includes ST-T changes, low voltage QRS complexes (if pericardial effusion is present) or LV hypertrophy with LA abnormality. The presence of AF in left-sided involvement often suggests an unfavourable prognosis. Echocardiography reveals increased endocardial echo-reflectivity, preserved systolic function, apical obliteration, enlarged atrium, pericardial effusion of varying sizes and Doppler ultrasound evidence of mitral regurgitation. Cardiac catheterization reveals pulmonary hypertension, LA hypertension and reduced cardiac index [33]. Finally, EMF with bi-ventricular is more common than isolated RV or LV involvement. Typical clinical manifestations resemble that of EMF with RV involvement although a murmur of mitral regurgitation often indicates LV involvement. In patients with extensive LV involvement, severe pulmonary hypertension is present although RV-findings are the predominant mode of presentation. Approximately 15% of patients will experience systemic embolization and only 2% will exhibit infective endocarditis [33].
Clinical evaluation: Clinical evaluation of EMF is based on a set of echocardiographic criteria, which has also been useful in staging the disease, studying its progression and comparing different epidemiological studies [74,75]. The criteria consists of six major criteria and seven minor criteria in which the presence of two major, or one major and two minor criteria establishes a diagnosis. The criteria has a scoring system with a score assigned to each criteria in which the total score indicates disease severity. A score < 8 indicates mild EMF; 8-15 moderate disease; and >15 severe disease [74].
Major Criteria
- Endomyocardial plaque > 2 mm thickness; score: 2
- Thin (≤ 1 mm) endomyocardial patches affecting > 1 ventricular wall; score: 3
- Obliteration of the RV and/or LV apex; score:4
- Thrombi or spontaneous echo contrast without severe ventricular dysfunction; score: 4
- Retraction of the RV apex (RV apical notch); score: 4
- AV valve dysfunction due to adhesion of valve apparatus to the ventricular wall; score: 1-4 depending on the severity of regurgitant lesion.
Minor Criteria
- Thin endomyocardial patches localized to single ventricular wall; score: 1
- Restrictive flow pattern across AV valves; score: 2
- Pulmonary valve diastolic opening; score: 2
- Diffuse thickening of anterior mitral leaflet; score; 1
- Enlarged atria with normal sized ventricles; score: 2
- M movement of the IVS and flat posterior wall; score: 1
- Enhanced density of the moderator or other intraventricular bands; score: 1
Endomyocardial biopsy is diagnostic but false negatives cannot be ruled out because of patchy involvement of the myocardium. The presence of systemic emboli may complicate myocardial biopsy contraindicating its use in left-sided myocardial biopsy [33,36]. In endemic areas, differential diagnosis should be made from right-heart disease, DCM, tuberculous pericarditis and constrictive pericarditis and thus supporting the need for echocardiographic assessment. History of rheumatic fever, evidence of mitral stenosis of aortic valve involvement favours the diagnosis of right heart disease but pure mitral insufficiency may be difficult to distinguish from left EMF when fibrosis and endocardial thickening affect predominantly the valve tissue. Right heart disease and EMF may co-occur in some patients [76].
Management: Pharmacological management of EMF consists of ameliorating acute disease and treatment of symptomatic HF [33,36]. In resource constrained settings where EMF is endemic, management of symptomatic HF includes HF medications (diuretics, angiotensin converting enzyme – inhibitors [ACE-I]) in combination with aspirin or anticoagulants [9]. Patients with advanced disease require larger doses and frequent hospitalization for invasive procedures to alleviate effusion and control arrhythmias. Oral corticosteroids have no or little influence on the naturel course of EMF and their use is not supported [77]. Management of ascites relies on frequent evacuation of fluid by paracentesis [78]. Patient with AF and/or thrombus on echocardiography require standard anticoagulation therapy. Diuretics are effective in the early stages of the disease to control HF but lose effectiveness with advanced ascites.
In patients with advanced EMF, surgical decortication with AV valve replacement on affected side is the choice treatment [79]. Surgical intervention increases survival and quality of life relative to medical therapy but should be performed prior to irreversible cardiac or hepatic damage [80]. Surgical intervention consists of conservative endocardiectomy and valve replacement or repair, resulting in improve hemodynamics with associated reduction in filling pressures, increased cardiac output and normalized angiographic appearance. However, operative mortality remains high (15 to 25%) but may be lower is valve replacement is unnecessary [81]. Relative contra-indications for surgery in resource-constrained settings include chronic ascites, extreme cachexia, chronic pulmonary embolism, extensive endocardial fibrosis or calcification, impaired myocardial function and extreme shortening of leaflets for anticipated valve replacement [82].
Despite treatment, EMF patients have ominous prognosis, which depends on the extent and the distribution of the disease within the various cardiac chambers and valves. The disease is progressive but with varying time course of decline [83]. Since a majority of patients have advanced disease at diagnosis, survival post-diagnosis is relatively short, about two years since symptom onset [84-86].
Hypereosinophilic heart disease
Prevalence: Hypereosinophilic syndrome (HES) is a heterogeneous group of rare haematological disorders often characterized by an unexplained and sustained blood eosinophilia (eosinophil counts > 1,500 per mm3 for ≥ 6 months) without any other known secondary cause (such as parasitic or allergy) and with the evidence of organ involvement [87]. Dermatological, pulmonary and gastrointestinal involvement are more common although cardiac involvement is the major source of morbidity and mortality [88]. Earlier studies reported up to 84% of HES patients have signs and symptoms of cardiac disease while recent studies suggest a lower frequency ranging between 40% and 50% [89,90]. The prevalence of HES is unclear although Spry reported a rate of 1 case in 200,000 people [91]. The disease has a male preponderance and tends to occur between the second and the fifth decade of life [87]. The most characteristic cardiac abnormality in HES is EMF, first described in 1936 by Loeffler, who termed it “fibroplastic parietal endocarditis with blood eosinophilia” [92]. It is a relatively rare conditions and an uncommon cause of RCM. Several types of cardiac damage may manifest in the context of eosinophilia toxicity in cardiac tissues, which range from acute MC to EMF [36]. HES patients may also develop thrombosis particularly in the cardiac ventricles but also occasionally in deep veins. Due to the rarity of HES, specific management guidelines of cardiac and thrombotic complications of HES are lacking [93].
Aetiology: Numerous diseases may be responsible for eosinophilia but not all cause clinically significant eosinophilia. Table 1 provides a summary of the most common aetiologies of chronic eosinophilia.
Table 1. Common aetiologies of chronic Eosinophilia
Aetiologies |
Specific Diseases |
Reactive eosinophilia |
Drugs (hypersensitivity): anticonvulsants, non-steroidal anti-inflammatory drugs; antimicrobial agents, sulphonamides.
Infections: helminths, HIV, human T-lymphocyte virus I, tuberculosis.
Allergic diseases |
Systemic diseases |
Crohn’s disease, Churg-Strauss syndrome, Wegener’s granulomatosis, polyarteritis nodosa, rheumatoid arthritis, cholesterol crustal embolism |
Malignancies |
Hodgkin lymphoma, non-Hodgkin lymphoma, acute leukaemia, systemic mastocytosis |
Hypereosinophilic syndrome |
Lymphocytic variant, myeloproliferative variant |
Source: Seguela et al. [36]
Reactive eosinophilia is a consequence of the use of some drugs such as anticonvulsants non-steroidal anti-inflammatory drugs, antimicrobial agents, sulphonamides, which trigger an abnormal production of eosinophils. Eosinophilia may also be the sole manifestation of drug-induced hypersensitivity reaction. Drug rash with eosinophilia and systemic symptoms syndrome refers to drug-induced eosinophilia with morbilliform eruption and severe tissue damage [94]. However, the withdrawal of the culprit drug often leads to normalization of eosinophil count within 7-10 days [87]. Vaccination of smallpox or diphtheria/tetanus/pertussis vaccines have also been reported to lead to eosinophilic MC [95,96]. In the presence of significant eosinophilia, immediate initiation of an empirical anti-helminthic drug therapy is recommended. Other aetiologies are mainly due to systemic diseases, malignancies and HES [36].
Natural course and pathophysiology: Cardiac involvement is one of the most frequent manifestation of sustained eosinophilia [87,97] often characterized by fibrosis that obliterates the ventricles with EMF as the ultimate form of eosinophilic cardiac disease [92]. Similar to EMF, the natural cause of cardiac pathology in HES has traditionally been divided into three chronological stages: eosinophilic infiltration, thrombosis and fibrosis [98,99].
Eosinophilic infiltration stage: Typical characteristics of the initial (eosinophilic infiltration or the acute necrotic) stage is eosinophilic endomyocarditis with eosinophilic and lymphocyte infiltration [93]. Eosinophils invade cardiac tissue, degranulate and release toxic cationic proteins inducing necrosis and apoptosis although patients do not generally have cardiac symptoms and may only present with non-specific signs [100]. Clinical and in vivo evidence of eosinophilic MC is infrequent while in autopsy series accounts for up to 0.5% [101]. Electrocardiography (ECG) abnormalities include sinus tachycardia, non-specific ST-segment changes or conduction delays but not clinically significant. Echocardiography abnormalities include increased LV wall thickness due to interstitial myocardial oedema. Endomyocardial biopsy (EMB) distinguished eosinophilic from other MC types. Histological analysis reveals eosinophilic infiltration of the endocardium and sub-endocardium interstitium, myocardial necrosis and sometimes eosinophilic granulomas [101,102]. Therapeutic target at this stage is rapidly lowering the eosinophil count to minimize myocardial necrosis [36].
In this initial stage, acute narcotising eosinophilic MC is the most severe form of the disease and is fatal without early diagnosis and institution of appropriate treatment [93,103]. Patients present with symptoms of acute HF or cardiogenic shock. Patients may exhibit conduction abnormalities, diffuse ST segment elevation and elevated troponin levels mimicking acute myocardial infarction, LV systolic dysfunction with wall motion abnormalities [104,105]. At this stage, cardiac magnetic resonance imaging (CMRI) is efficient in detecting endomyocardial involvement, particularly late gadolinium enhancement (LGE) that shows extensive eosinophilic infiltrates and at times patchy distribution [106]. Eosinophilic infiltration due to hypersensitivity mechanism may manifest with normal or mildly elevated peripheral eosinophil counts [36]. However, about half of patients do not exhibit eosinophilia at disease onset, which occurs due to the migration of circulating eosinophils into the tissue while bone marrow cannot respond immediately with increased production [107]. Thus, repeated white blood examinations are necessary in patients with initially absent eosinophilia. Since corticosteroid therapy is the first-line treatment for eosinophilic MC because of its inhibition of the degranulation of eosinophils limiting myocardial necrosis but its efficacy remains controversial [108,109]. Treatment of symptoms of HF rests on HF conventional drugs while in fulminant HF may require mechanism support [36].
Thrombotic stage: Persistent eosinophilic activation leads to the second (thrombotic) stage of the disease, characterized by the formation of mural thrombi along the damaged myocardium due to tissue necrosis often involving both ventricles, the ventricular flow tracts and the sub-valvular regions [36,93]. Eosinophilic proteins bind to thrombomodulin impairing the anticoagulant properties of the endothelial membrane leading to AV valvular incompetence [110,111]. Disruption of the normal anticoagulant endothelial lining exposes the von Willebrand factor (vWf), collagen and tissue factor. The vWf and collagen bind platelets and tissue factor binds activated factor VII to initiate the biochemical reactions that generate a fibrin thrombus [112,113]. In HES patients, the stimulation of fibrin formation by tissue factor may be especially important because eosinophils contain tissue factor in their specific granules that can induce the endothelium to express it [114,115]. Recent evidence suggests that eosinophil granule proteins can activate tissue factor XII and platelet and mononuclear cells from HES patients may have enhanced pro-coagulant activity [116-118]. Thus, blood hyper-coagulability may also contribute to the pathogenesis of thrombosis in HES patients. Thrombotic events occur in 14 to 29% of adult patients with idiopathic eosinophilia legitimizing the use of oral anticoagulation or antiplatelet therapy to prevent thrombi formation at the stage of eosinophilic MC but evidence of the effectiveness of these therapies are lacking [87,90,119]. Cardiac chambers have a relatively static flow at the endomyocardial surface particularly in the ventricular apices and becomes more static in areas of hypokinesia. Stasis is a major stimulus for thrombosis since it allows activated clotting factors o accumulate to thrombotic concentrations [93].
Fibrotic stage: In the final fibrotic stage, fibrosis replaces thrombus formed on the denuded myocardium corresponding to scarring of the endomyocardium. Fibrosis is an irreversible damage involving both the RV and the LV, and may include sub-valvular apparatus of bot mitral and tricuspid valves [120-122]. Diagnosis of cardiac involvement in HES patients often is often made in this final fibrotic stage when they present with scarring of the chordae tendineae and endocardium resulting in RCM or DCM with an ominous prognosis sometimes accompanied by progressive valvular incompetence [123]. Valvular pathology frequently observed in HES patients include dysfunctional, regurgitant AV valves due to restriction of valve leaflet mobility but aortic and mitral stenosis are less common [124]. Gottdiener et al. echocardiographic series reported mitral regurgitation in 43% of HES patients [125]. The accumulation of thrombofibrotic material between the mural endocardium of the LV free wall and the ventricular aspect of the posterior mitral leaflet limited posterior mitral leaflet motion [126]. EMF may also be responsible for altered cardiac conduction system leading to severe ventricular arrhythmias [127].
Clinical presentation: Patients with HES and cardiac involvement usually present with signs and symptoms of HF, intracardiac thrombus, myocardial ischemia and arrhythmias but rarely pericarditis [90,128]. In a prospective study of 26 HES patients with cardiac involvement and a review of literature, Parrillo et al. reports the most common presenting symptom in literature is dyspnoea occurring in 60% of the patients. In 75% of 55 patients, who could be evaluated, exhibited signs and symptoms of HF and 4% had evidence of pericarditis [90]. In the prospective part, common symptoms were dyspnoea (42%), chest pains (2%), cough (12%), palpitations (8%) and embolic events (4%) [90]. In subsequent evaluation, signs and symptoms included mitral regurgitation (42%), HF (38%), aortic regurgitation (4%) and aortic stenosis (4%). Although myocardial infarction is a rare complication, it could manifest as a consequence of embolic event secondary to endomyocardial fibrosis and thrombus in the LV outflow tract [129]. Earlier description of risk factors for cardiac HES cardiac disease included male sex, HLABw44 positivity, splenomegaly, thrombocytopenia, elevated serum vitamin B12, dysplastic eosinophils and the presence of abnormal early myeloid precursors [99]. However, recent evidence suggests that these features are characteristics of myeloproliferative variant of HES [130].
Clinical evaluation: Chusid et al. formulated the definition of HES with strict diagnostic criteria [87]:
- Peripheral blood eosinophil count > 1500 cells/mm3 for at least 6 months duration;
- Signs and symptoms of end-organ involvement with eosinophil tissue filtration; and
- Exclusion of known secondary causes of eosinophilia.
These criteria involves clinical evaluation of HES cardiac disease using a combination of ECG, echocardiography, CMRI and EMB. Typical but non-specific ECG abnormalities include T-wave inversion, LA enlargement, LV hypertrophy, incomplete RBBB and left axis deviation [90]. Other reported ECG abnormalities may include ventricular premature complexes, poor R wave progression, non-specific ST-T wave changes, and first-degree AV block [131]. Although useful, ECG only detects changes related to cardiac pathology especially LV hypertrophy but does not reveal abnormalities specific for HES [93]. Echocardiography has a long history in the evaluation of cardiac disease on HES patients. Different modalities, 2D, transthoracic, transesophageal, and contrast echocardiography play complementary roles in clinical evaluation of HES. Contract echocardiography delineates LV shape and quantifies LV hypertrophy [132]. Classical echocardiographic findings include endomyocardial thickening, bi-ventricular apical thrombus formation, and posterior mitral leaflet involvement [89] with progressive features of restrictive physiology with regurgitation of the AV valves secondary to sub-valvular injury [111].
Recently, CMRI is emerging as a highly useful non-invasive modality for the diagnosis of cardiac disease in HES patients. It is sensitive and specific for detecting ventricular thrombi than echocardiography and LGE-CMRI is able to detect myocardial fibrosis and inflammation [133,134]. LGE due to fibrosis is more intense than LGE due to inflammation [133]. Despite improvement in non-invasive cardiac imaging modalities in the diagnosis of HES cardiac disease, EMB remains the diagnostic gold standard. Serial myocardial biopsies provide information on the clinical course of cardiac disease and response to treatment. Common EMB findings include fibrotic thickening of the endocardium, mural thrombosis and fibrinoid changes, thrombosis and inflammation of small intramural coronary vessels and, sometimes, eosinophil infiltration into the myocardium and sometimes endocardium [98]. However, high resolution CMRI permits tissue characterization rendering this technique promising and practical to follow and potentially diagnose HES cardiac disease [93].
Clinical management: Due to the rarity of cardiac disease in HES, clinical guidelines for its management are lacking. Nevertheless, there is a role for both medical and surgical therapies in improving the quality of life in HES patients with cardiac involvement by reducing eosinophil count [7]. Several case reports demonstrate favourable outcome with the use of corticosteroid and cytotoxic drugs such as hydroxyurea. Interferon may also be beneficial as adjunctive therapy in refractory patients. Anticoagulation also can reduce the thrombotic burden [135]. Novel targeted therapies have been developed to target eosinophilic-associated disorders including treatment for HES and eosinophilic MC, which precedes the development of CM. Interleukin-5, produced by the TH2 helper cells, is the main cytokine responsible for eosinophilic differentiation, survival and activation. Therefore, it is the natural target for molecular therapies. Two monoclonal antibodies (mepolizumab and reslizumab), which bind interlukin-5 have been developed and tested in various eosinophilic-associated disorders with results showing its safety and efficacy in reducing eosinophilia but with modest clinical benefits. Mepolizumab was evaluated in HES patients and met the primary endpoint if steroid reduction but has not been approved for the treatment of HES [136]. Many biological targeting eosinophilic surface receptors and soluble mediators associated with eosinophilic inflammation are under investigation [137].
Endocardial fibroelastosis
Prevalence: Endocardial fibroelastosis (EFE or foetal endocarditis) is a rare and poorly understood endomyocardial disease with typical restrictive HF manifestations and cardiac arrhythmias predominantly encountered in sub-Saharan Africa. The term EFE was introduced in 1943 to describe a pronounced fibroelastic thickening of the ventricular endocardium presented as unexplained HF in infants and children [138]. Originally, EFE had been classified as diffuse idiopathic (primary) or secondary to other myocardial diseases such as viral infections, MC, metabolic disorders, inherited diseases, and congenital malformations [139,140]. However, the existence of primary EFE has been questioned as whether it is a distinct clinical entity or a consequence of dilated, infectious of inflammatory CM. Indeed it is argues that the term primary EFE suggests that it presents a common final pathway of many myocardial diseases. Consequently, the most recent ESC and AHA classification of CMs have excluded EFE as a type of CM; instead, include both EMF and EFE within the clinical spectrum of RCM [4-6] although some studies include EFE as an aetiology or consequence of DCM [139,140].
Despite the controversy of primary EFE-associated CM, cases of EFE have been reported since the 1960s. In 1964, a study in the U.S reported an incidence of 1:5,000 births [141]. In 1978, the reported incidence rate of EFE was 1-2% of all congenital heart diseases. However, in a recent review of medical records, echocardiograms, explanted hearts and microscopic slides of 53 paediatric patients with clinical diagnosis of DCM, Seki et al. found primary EFE in 25% of paediatric patients, suggesting that the entity is not are rare as previously thought [141]. The study reports that endocardial thickening in primary EFE is distinctive and characterized by thick and parallel oriented endocardial elastic fibres resembling the thick elastic lamina of the aorta and are structurally and biochemically different from the smaller, poorly oriented fibres observed in the secondary forms of EFE [141]. In a retrospective Finnish study reviewing medical records of patients aged birth to 20 years from 1980-1991 enrolling 118 having idiopathic CMs, 62 had DCM. Of the 62, 12 (19%) were diagnosed with EFE CM [142]. All the patients with EFE were younger than 2 years at diagnosis (median 4.8 months) and 8 (67%) were female [142]. Fewer cases of EFE have been described in which older children, adolescents or even adults presenting with cardiac failure have been diagnosed with EFE [143].
Aetiopathogenesis: The aetiopathogenesis of EFE is not fully understood but it is suggests to develop as a result of inflammatory, mechanical or hereditary factors. Primary or secondary EFE often mimics DCM [141]. More commonly, EFE manifests in association with other structural cardiac abnormalities, including left-sided obstructive lesion such as aortic stenosis and anomalous coronary artery arising from pulmonary trunk. It is likely that fibroelastosis represents the end-stage pathological phenotype of a heterogeneous group of conditions rather than a single disease entity [5]. Possible aetiopathogenic agents include viral infections with coxsackievirus, adenovirus, parvovirus, and mumps virus [5,140,144]. EFE may also be associated with metabolic and storage disorders such as primary deficiency of carnitine and respiratory chain abnormalities [145,146]. Recently, EFE has been recognised in foetal and postnatal life, in association with maternal antibodies (anti-RO and anti-La) in children with and without congenital complete heart block [147,148].
Some authors have also described hereditary element in EFE. Arola et al. described two families that had patients’ phenotype that was typically EFE or DCM without EFE suggesting that EFE might be secondary to heart dilatation in a predisposed individual [142]. Chen et al. reported nine patients with familial EFE in four families inherited in X-linked recessive, autosomal dominant and autosomal recessive fashion [149]. Recent reports have associated mutations in the tafazzin (TAZ/G4.5) gene located on Xq28 with familial X-linked EFE and Barth syndrome, reported to result in structural changes in the foetal heart as early as 18 weeks of gestation [150,151]. In a summary, current evidence suggests that primary EFE is hereditary, occurs with no other demonstrable cause of unexplained HF while secondary EFE is associated with aortic stenosis or atresia and includes coarctation of the aorta, ventricular septal defect, anomalous origin of left coronary artery from the pulmonary artery, myocardial injury from any cause, and metabolic or carnitine deficiency.
Pathophysiology: Typical pathological characteristics of EFE are diffuse endocardial thickening and myocardial dysfunction. Endocardial thickening may be the consequence of persistent and increased wall tension in the ventricles possible secondary to injured myocardium, mitral regurgitation or both [140-144]. EFE is sporadic but familial cases have been reported [150,151]. The suggestions that EFE might have a viral aetiology stems from the observation of the similarity of its clinical presentation to that of chronic MC. Similarities include findings of MC or myocardial fibrosis in EFE patients, a higher incidence of EFE following epidemics of coxsackievirus B infection, persistence of viral infection in EFE patients and experimental production of EFE in animal models by viral infection of the myocardium, demonstration of persistent viral infection with molecular studies, and experimental production of the disease in animal models by viral infections of the myocardium [140,143,144].
EFE CM has a phenotypic resemblance to DCM. Patients with dilated EFE have a markedly enlarged globular heart, mainly involving the LV and the LA. The LV endocardium is opaque, glistening, milky white and diffusely thickened (1-2 mm) most markedly in the outflow tract [152]. Papillary muscles arise more superiorly on the ventricular wall with thickened and shortened chordae tendineae. Flattened papillary muscles and trabeculae carneae partially incorporated in the fibrotic processes exert an undesirable lateral traction on the chordae tendineae and mitral cusps resulting in faulty leaflet opposition [153,154]. EFE patients have a thickened endocardium although the ventricular walls (myocardium) is within the reference range. Endocardial thickening may extend to the LA, RV and RA [152]. The less common contracted type of primary EFE due to hypoplastic HF has a relatively hypoplastic is normal LV size [152]. The RA, LA and RV are markedly enlarged and hypertrophied with minimal or no endocardial sclerosis. Secondary EFE, associated with cardiac malformations, may arise due to cardiac hypertrophy and consequent imbalance in the myocardial oxygen supply and demand relationship. The resultant fibroelastotic thickening is often focal and less severe [143].
EFE-associated HF becomes progressive with age resulting in death within the first six months of life. In a sub group of patients who survive from a few months to several years, a more chronic phase of the disease ensues. These patients may respond to the conventional HF medication. A variable cyclical clinical course may ensue, with HF recurrences due to respiratory or other inter-current infections or to the progression of the disease. Remissions can occur with intensification of medical therapy although cardiac arrhythmias can be the main presenting problem [143].
Clinical evaluation: Often, EFE presents in infancy and clinical presentation are typically indistinguishable from those seen in DCM patients. In many cases, EFE progresses to end-stage HF and death. The onset of EFE may be acute producing cardiogenic shock or sudden cardiac death in infancy, a history of frequent or recent respiratory tract infection, episodes of severe sudden abdominal pain may indicate coronary insufficiency, and the contracted form of EFE presents with features of left-sided obstructive disease and acute LV failure [143]. Diagnosis methods for EFE rely largely on evidence from earlier studies with no recent guidelines despite improvement in non-invasive cardiac imaging modalities. Recent studies are warranted to clarify and refine diagnosis of EFE heart disease. At present, diagnosis rests on histological and the pathological hallmarks of the deposition of collagen and elastin, ventricular hypertrophy and diffuse endocardial thickening [141-143].
Typical ECG abnormalities in primary EFE include conduction defects (prolonged PR interval, intermittent heart block and wide QRS complexes [155]. Echocardiographic changes may include increased LA and LV dimensions, depressed ejection fraction, abnormal mitral valve motion, dense echogenicity along the endocardium of the LV, globular LV shape, and varying degree of mitral regurgitation [141-144]. Echocardiogram is able to demonstrate vigorous systolic function but substantial diastolic dysfunction on the LV. There is mild endocardial brightening of the anterior septum, anterior wall or papillary muscles [156]. However, echocardiography may not be a sensitive diagnostic test since neither echo brightness not LV dimension can reliably detect the presence of EFE [143].
The diagnostic role of CMRI has been recently highlighted in detecting the presence of EFE [156,157]. CMRI using perfusion and myocardial LGE can be a useful adjunct in establishing diagnosis. EFE gives the endocardial surface rim of hypointense signal in the perfusion sequences and a rim hyperintense signal in the myocardial LGE sequences [157]. CMRI demonstrates the pathognomonic features of endocardial fibroelastosis and can identify additional complications such as intracardiac thrombus [156]. EMB may be used on cases where diagnostic is unclear. However, EMB has its risks especially in infants and is not essential to make a diagnosis in the majority of affected babies. Usually, EMB reveals invasion of the endocardium and sub-endocardium by fibroelastic tissue [158].
Management: EFE lacks directed therapy due to the rarity of the disease and the current recommended management follows that of HF in general such as targeting HF symptoms with optimization of volume status, managing rate control (in case of AF with rapid ventricular rates) and risk stratification for conduction abnormalities for candidacy of implanted devices [159]. Medical management of EFE is essentially the same to that of chronic HF and its acute exacerbations are the consequence of respiratory infections. Early and prolonged digoxin therapy continued for several years since the disappearance of symptoms. Cessation of digoxin may result in acute HF even when cardiac size has normalized [159]. Other management strategies for acute HF and precipitating factors such as anaemia may require attention. Anticoagulation may be required if the patients has thromboembolic complications. Case reports have also cited resolution of antenatally diagnosed EFE associated with positive maternal antibodies (anti-Ro and anti-La) with corticosteroid therapy. Cardiac transplantation may be required for end-stage HF [160,161].
The present meta-analysis sought to evaluate the main diagnostic features of CM due to endomyocardial diseases. The search for relevant studies was performed on PubMed, Cochrane and Google Scholar from inception to June 2019. The key words and medical subject heading (MeSH) terms of endomyocardial diseases and cardiomyopathy along with individual names of causative diseases (endomyocardial fibrosis, hypereosinophilic heart disease, and endocardial fibroelastosis). Reference lists of articles that fulfilled the inclusion criteria were scanned to identify additional articles missed by the online search. There was no limitation to language or publication period. However, the studies has to meet the following inclusion criteria: (i) enrolled patients with EMF, HES and/or EFE; (ii) were diagnosed using serum, ECG or non-invasive cardiac imaging; and (iii) reported the findings of the diagnostic tests. Case reports, editorial and review articles were excluded. The primary outcomes assessed were diagnostic findings from different tests.
Only data published in the included studies were used as to form the final dataset for analysis. In the primary analyses, diagnostic features for continuous outcomes are expressed as event rate with 95% confidence intervals (CIs). Analysis employed random effect model and fixed effect model to calculate pooled outcomes of diagnostic features. The inconsistency parameter (I2) was used to test for heterogeneity. Statistical significance for diagnostic features was defined by P values < 0.05 and heterogeneity as P values < 0.10 or I2 > 25%). When heterogeneity was present, an explanation was sought in terms of methodological quality and different clinical characteristics. When no clinically significant differences between studies would be identified, the results of studies were pooled together using the random effect model.
Study characteristics
The online search yielded 198 potential references, out of which only seven articles meeting the inclusion criteria were included [49,89,90,125,141,162,163]. The seven articles were published between 1979 and 2015 [90,162,163]. A greater majority of the excluded articles were case reports of one to three patients while few others included patients with mixed conditions making data extraction of patients of interest difficult. Table 2 provides a summary of the key characteristics of the studies included in the present meta-analysis. In total, the seven studies enrolled 235 patients diagnosed with CM due to endomyocardial disease. The mean age varied significantly from 7.5 days to 45±17 years. EFE patients were younger (7.5 days to 10.1 months) while EMF and HES patients were relatively older (18±11 to 45±17), which mirrors the common age of presentation of the respective underlying endomyocardial disease. Three studies studies HES patients, three studied EFE while the remaining one studied EMF [49,89,90,125,141,162,163]. All the seven studies, except one that evaluated serum tests (antibody reactivity), studied echocardiographic features of endomyocardial diseases.
Table 2. Summary of included studies
Author (year) [Ref #] |
Patient Selection |
Mean Age |
Cause |
Diagnostic Test |
Summary of Key Findings |
Parrillo [90] |
26 patients with HES followed for 9 years |
NA |
HES |
2D-Echo |
Dyspnoea (42%); chest pain (27%); HF signs (38%); MR (42%); cardiomegaly (35%); T-wave inversion (35%); echo abnormalities (82%) |
Gottdiener [125] |
21 consecutive patients with the idiopathic HES |
39±12 |
HES |
2D-Echo |
MR (9); LV wall thickness (9); PE (6); intraventricular thrombus (10); |
Ommen [89] |
51 patients with idiopathic HES from medical records based on Mayo clinic diagnostic index 1967-1995 |
45±17 |
HES |
Echo |
Endocardial thickening (6); LV apical thrombus (12); RV apical thrombus (10); PML involvement (10); LV hypertrophy (5) |
Schmaltz [162] |
8 normal infants and 7 infants with EFE based on clinical picture, cardiac catheterization and angiography |
3.29 months |
EFE |
Echo |
EFE have higher dimensions of LV cavity (D) than normal infants: Dmax (35.9±7.3 vs. 20.7±2.6), Dmin (33.5±7 vs. 12.7±2.5 mm); reduced shortening fraction (11,7±4,6 vs. 35.7±7.5) |
Dogan [163] |
22 newborns (17 boys, 5 girls) with EFE due to aortic stenosis 2008-2014 |
7.5 days |
EFE |
Echo |
Mean EF (34±13); FS (17±6.5); mortality rate (n=7; 31%); reduced peak-to-peak aortic valvular gradients pre and post-valvuloplasty (56±32 and 22±15 mmHg) after 38±20 months |
Mocumbi [49] |
56 patients with EMF on echo and eosinophil count >1.5x106 and 10 healthy controls |
18±11 |
EMF |
Serum screening |
IgG reactivity against myocardial protein stronger in EMF than healthy controls (30/56 53.6% vs. 1/19, 10%) and IgM (11/56, 19.6% vs 0/10, 0%). EMF has greater frequency and reactivity of IgG against myocardial proteins of molecular weight |
Seki [141] |
52 pediatric heart transplant DCM patients with primary EFE reviewed from medical records |
10.1 months |
EFE |
2D echo |
14 hearts with gross and microscopic findings of primary EFE. Patients with EFE were younger than DCM; greater LV wall and endocardial thickness greater in EFE with mitral valve and papillary muscles showing characteristic findings |
DCM: Dilated Cardiomyopathy; EFE: Endocardial Fibroelastosis; Echo: Echocardiograph; EMF: Endomyocardial Fibrosis; HES: Hypereosinophilic Syndrome; MR: Mitral Regurgitation; PE: Peripheral Embolism
Meta-analysis : Due to the lack of clinical trials on CMs due to endomyocardial diseases, there was insufficient data to enable pooled analysis to determine the most common pathologic features that can be useful to support diagnosis. Only three studies enrolling HES patients could be pooled despite a small sample of patients enrolled in the individual studies. Pooled analysis of three studies revealed 32 out of 98 HES patients had echocardiographic evidence of LV wall thickness, translating into an event rate of 36.4%, 95% CI 9.4% to 76% (Figure 1) [89,90,125]. There was significant heterogeneity (I2 = 92%) noted between the three studies, which could not be explained in terms of clinical characteristics at baseline or methodological quality. Thus, a random effect model was used to perform pooled analysis. Pooled data from two studies revealed 14 out of 72 HES patients had peripheral emboli (event rate: 20.5% 95% CI: 10.9 to 35.2%; Figure 2), 19 out of 72 had posterior mitral leaflet (event rate: 29.3% 95% CI: 12.1% to 55.4%; Figure 3), and 32 out of 72 had intraventricular thrombus (event rate: 44.5 95% CI: 33.4 to 34.8%; Figure 4) [89,125]. Parrillo et al. [90] reported that 88% of HES patients exhibit echocardiographic abnormalities and 38% had signs and symptoms of HF.
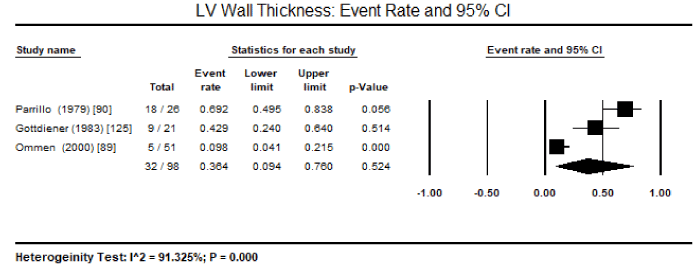
Figure 1. Event rate and 95% CI for LV wall thickness
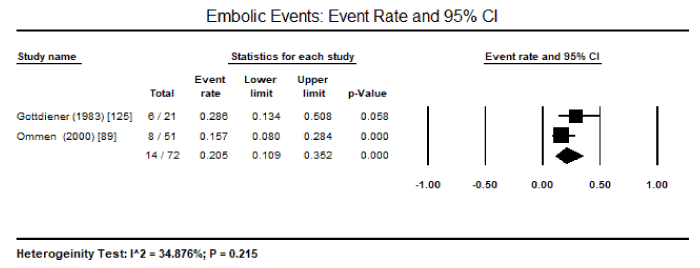
Figure 2. Event rate and 95% CI for peripheral emboli
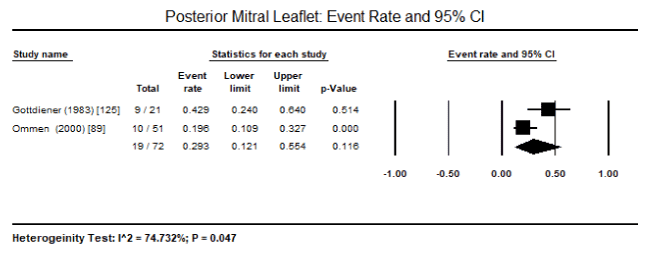
Figure 3. Event rate and 95% CI for posterior mitral leaflet
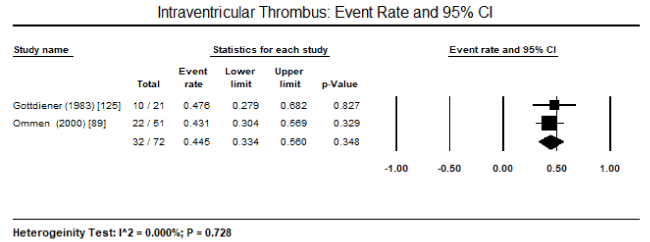
Figure 4. Event Rate and 95% CI for intraventricular thrombus
Systematic review: Calculating pooled findings on diagnostic features for EFE and EMF was not possible because of insufficient data or incompatible data provided for diagnostic parameters of interest. Nevertheless, the individual studies on EFE and EMF communicated important information about the diagnosis of EFE and EMF.
Based on a review of medical records, echocardiograms, explanted hearts, and microscopic slides, Seki et al found EFE was more prevalent that earlier believed manifesting in 25% of 52 infants diagnosed with DCM [141]. Patients with EFE had evidence of significant LV wall and endocardial thickness than DCM patients, with mitral valve and papillary muscles showing characteristic findings.
In 22 newborn diagnosed with EFE associated with aortic stenosis, Dogan et al. reported he LV systolic function varied, with 90% of patients with LVEF < 55% [163]. The mean LVEF was 34±13% and fractional shortening of 17±6.5%. Further, EFE demonstrated a higher early mortality rate (31%) despite improvement in peak-to-peak aortic valvular gradients pre (56±23 mmHg) and post (22±15 mmHg) valvuloplasty.
Schmaltz et al, compared seven EFE infants (mean age: 3.29 months) with eight normal infants (mean age = 3.2 months) matched to age and surface area [162]. The study reported EFE infants has higher maximum and minimum dimensions of LV cavity (Dmax: 35.9±7.3 vs. 20.7±2.6; Dmin: 33.5±7 vs. 12.7±2.5 mm) and significantly reduced fraction shortening (11,7±4.6% vs. 35.7±7.5%).
Mocumbi et al. evaluated the possibility of endocardial lesion being a result of immune response against myocyte on 56 EMF patients by assessing the frequency of circulating anti-myocardial antibodies [49]. The study reported stronger IgG reactivity against myocardial proteins (53.6%) compared to healthy controls (10%). Although IgM reactivity was weaker, it was still greater among EMF patients (19.6%) than healthy controls (0%) suggesting the role of autoimmunity.
To date, the diagnosis of CMs due to EMF and HES is based on criteria developed in the past based on eosinophilic count > 1,500 cells/mm3persistign for >6 months and evidence of end-organ involvement without any other known cause of eosinophilia while the criteria for EFE diagnosis remains elusive. The description of CMs due to endomyocardial diseases within the morphological and structural features of RCM may have limited research on its natural course, diagnosis and management. In the course of study search, the present systematic review and meta-analysis attests to the scarcity of studies on endomyocardial diseases, undermining a precise understanding of this disease entity and its role on the pathogenesis of CM. Despite the lack of data, the seven studies included for analysis provide important information about pathological features of endomyocardial disease that might support diagnosis although there is a need for additional large-scale prospective clinical trials to clarify these findings as well as their diagnostic value.
In the present analysis, data based only on HES patients could be pooled. In addition, very few diagnostic features could be studied because of heterogeneity in diagnostic parameter assessed. Pooled data from three studies revealed LV wall thickness on echocardiography is a common feature in HES patient, occurring in 36.4% of the patients. Other common features included peripheral emboli in 20.5% of the patients, posterior mitral leaflet in 29.3% of the patients, and intraventricular thrombus in 44.5%. These findings suggest that echocardiography is an important non-invasive diagnostic modality for evaluating cardiac manifestations of HES – thickening of postero-basal wall associated with impaired posterior mitral leaflet function resulting in mitral regurgitation and peripheral embolization and thrombus formation. The selection of the patients was based on criteria defined by Chusid and co-workers based on eosinophilic count and the exclusion of other known causes of eosinophilia [87]. The present findings are consistent with earlier reports of LV hypertrophy, endomyocardial thickening, bi-ventricular apical thrombus formation, posterior mitral leaflet involvement are classical echocardiographic features of HES suggesting restrictive physiology accompanied with regurgitation of AV valves [111,132]. Although ECG changes have been mentioned in literature, only the Parrillo study provided data on ECG changes in HES patients: LA enlargement, LV hypertrophy, ventricular premature complexes, poor R-wave progression, non-specific ST-T changes and first-degree heart block [90]. However, the role of ECG in diagnosis of HES is limited since it only provides evidence of cardiac pathology but does not reveal evidence of specific abnormalities associated with HES [132].
Besides echocardiographic findings in HES patients, there was no concrete evidence supporting diagnostic features of EFE and EMF from the available data and a meta-analysis was not feasible now because of marked heterogeneity of the available studies in terms of diagnostic methods, patient characteristics and outcome measures. EFE is a rare clinical entity and even a more rare cause of CM and its true incidence in the general population has remained unknown. Individual large-scale studies are difficult to conduct and thus the small sample size used by the studies included in the present meta-analysis, which result in the lack of statistical power to determine common diagnostic features and any significant differences. Unlike HES and EMF, which have well-defined diagnostic (selection) criteria, the criteria for inclusion of EFE patients need to be clarified. EFE is a disease of infants and adolescents that lacks diagnostic guideline as well as it is difficult to diagnose at initial presentation. Dogan et al. recruited newborn with EFE associated with aortic stenosis diagnosed based on patients’ demographic, echocardiographic and cardiac catheterization, and angiography findings . Diagnostic findings provided included LVEF and fractional shortening. Schmaltz et al. [162] recruited seven infants with EFE diagnosed based on clinical picture, cardiac catheterization and angiography [163]. Diagnostic outcomes provided included a comparison between EFE and normal infants based on LV cavity dimensions, fractional shortening. Finally, Seki et al. recruited 52 paediatric patients with explanted hearts diagnosed with DCM [141]. Diagnostic outcomes compared echocardiographic findings between DCM and EFE based on LV wall thickness, endocardial thickness and involvement of mitral valve and papillary muscles.
However, individual studies provided findings about pathological features that could assist in diagnosis of patients with EFE and EMF. Echocardiographic abnormalities are more common in HES patients (82%) compared to clinical, roentgenographic or electrocardiographic evidence of cardiac involvement (55%) [90]. Paediatric patients with EFE have significant LV wall thickness and endocardial thickness greater than that of DCM patients, as well as the involvement of mitral valve and papillary muscles [141]. In newborn with EFE secondary to aortic stenosis, systolic function is depressed and have a higher mortality rate (31%) [163]. Compared to normal infants, EFE infants have higher LV cavity dimensions and significantly reduced systolic function (fractional shortening). Finally, compared to healthy controls, EMF patients have a stronger IgG and a weak IgM reactivity with myocardial proteins of different loads, suggesting autoimmunity may play a role in the pathogenesis of EMF [49]. Whereas these findings suggest diagnostic feature that can be evaluated in the diagnosis of EFE, additional large-scale clinical trials or systematic review of case reports and case series may help to clarify their diagnostic role.
Endomyocardial diseases such as EMF, HES and EFE are rare but important causes of CM although they are underappreciated because they are described within the RCM phenotype under the structural-functional classification of CMs. EMF and HES are closely related diseases because they may share an eosinophilic involvement in their pathogenesis while in EFE, cardiac abnormalities (aortic stenosis or anomalous coronary artery arising from pulmonary trunk), infections and genetic may be involved in the pathogenesis. Clinical presentation is non-specific and diagnosis is not straightforward in all three endomyocardial diseases. The diagnosis of EMF includes two of the following major criteria: endomyocardial plaque >2 mm thickness; thin (≤ 1 mm) endomyocardial patches affecting >1 ventricular wall; obliterated RV and/or LV apex; thrombi or spontaneous echo contrast without severe ventricular dysfunction; retraction of the RV apex and AV valve dysfunction. The presence of two other minor criteria may also help to confirm diagnosis. The diagnostic criteria for HES is peripheral blood eosinophil count >1,500 cells/mm3 for at least 6 months duration; signs and symptoms of end-organ involvement with evidence of eosinophil tissue infiltration; and exclusion of other known causes of eosinophilia. Only EFE lacks specific diagnostic criteria although its pathologic hallmark is diffuse endocardial thickening accompanied by myocardial dysfunction. Clinical management depends on the underlying disease. Clinical management of EMF consists of ameliorating acute disease by corticosteroid therapy and the treatment of signs and symptoms of HF using standard HF medications. Management of HES consists of reducing eosinophil count either medically (using corticosteroid and cytotoxic drugs) or surgically. Management of EFE consists of HF medication with optimization of volume status and management of rate control. For a better understanding of the natural course of endomyocardial diseases, additional research may be beneficial to paint a clear picture of its pathogenesis, improve diagnosis and develop targeted treatment.
- Brigden W (1957) Uncommon myocardial diseases: the non-coronary cardiomyopathies. Lancet 273: 1179-1184. [Crossref]
- Goodwin JF, Gordon H, Hollman A, Bishop MB (1961) Clinical aspects of cardiomyopathy. Br Med J 1: 69-79. [Crossref]
- Corrado D, Basso C, Judge DP (2017). Arrhythmogenic cardiomyopathy. Circ Res 121: 784-802. [Crossref]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, et al. (2007) Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29: 270-276. [Crossref]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, et al. (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation. 113: 1807-1816. [Crossref]
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Trachtenberg BH, Hare JM (2017) Inflammatory cardiomyopathic syndromes. Circ Res 121: 803-818. [Crossref]
- Braunwald E (2017) Cardiomyopathies: an overview. Circ Res 121: 711-721. [Crossref]
- Grimaldi A, Mocumbi AO, Freers J, Lachaud M, Mirabel M, et al. (2016) Tropical endomyocardial fibrosis: natural history, challenges, and perspectives. Circulation 133: 2503-2315. [Crossref] PLoS Negl Trop Dis
- Bukhman G, Ziegler J, Parry E (2008) Endomyocardial fibrosis: still a mystery after 60 years. PLoS Negl Trop Dis 2: e97. [Crossref]
- Mocumbi AO (2012) Endomyocardial fibrosis: a form of endemic restrictive cardiomyopathy. Glob Cardiol Sci Pract 2012: 11. [Crossref]
- Mocumbi AO, Ferreira MB (2010) Neglected cardiovascular diseases in Africa: challenges and opportunities. J Am Coll Cardiol 55: 680-687. [Crossref]
- Celermajer DS, Chow CK, Marijon E, Anstey NM, Woo KS (2012) Cardiovascular disease in the developing world: prevalences, patterns, and the potential of early disease detection. J Am Coll Cardiol 60: 1207-1216. [Crossref]
- Mocumbi AO, Ferreira MB, Sidi D, Yacoub MH (2008) A population study of endomyocardial fibrosis in a rural area of Mozambique. N Engl J Med 359: 43-49. [Crossref]
- Jaiyesimi F (1980) The aetiopathogenesis of endomyocardial fibrosis: problems and promises. Tropical Cardiology 6: 113-120.
- Freers J, Mayanga-Kizza H, Ziegler JJ and Rutakingirwa M (1996) Echocardiographic diagnosis of heart disease in Uganda. Trop Doct 26: 125-128. [Crossref]
- Mckinney B (1975) Studies on the experimental production of endomyocardial fibrosis and cardiomegaly of unknown origin by dietary means. Am Heart J 90: 206-214. [Crossref]
- Nair DV (1971) Endomyocardial fibrosis in Kerala. Indian Heart J 23: 182-190. [Crossref]
- Kurian S, Nair DV (1980) Ecology of endomyocardial fibrosis in Kerala State. Indian Heart J 32: 156-162. [Crossref]
- Kutty VR, Abraham S, Kartha CC (1996) Geographical distribution of endomyocardial fibrosis in south Kerala. Int J Epidemiol 25: 1202-1207. [Crossref]
- Yin R (2000) Endomyocardial fibrosis in China. Chin Med Sci J 15: 55-60. [Crossref]
- Guimaraes A (1993) Natural history and current status in Brazil. In: Valiathan M, Somers K, Kartha CC, eds. Endomyocardial Fibrosis. Delhi: Oxford University Press 37-54.
- Bukhman G, Ziegler J, Parry E (2008) Endomyocardial fibrosis: still a mystery after 60 years. PLoS Negl Trop Dis 2: e97. [Crossref]
- Brockington IF, Edington GM (1972) Adult heart disease in western Nigeria: a clinicopathological synopsis. Am Heart J 83: 27-40. [Crossref]
- Falase AO (1983) Endomyocardial fibrosis in Africa. Postgrad Med J 59: 170-178. [Crossref]
- Connor DH, Somers K, Hutt MS, Manion WC, D’Arbela PG (1967) Endomyocardial fibrosis in Uganda (Davies’ disease), 1: an epidemiologic, clinical, and pathologic study. Am Heart J 74: 687-709. [Crossref]
- Shaper AG (1972) Cardiovascular disease in the tropics, II: endomyocardial fibrosis. Br Med J 3: 743-746. [Crossref]
- D’Arbela PG, Mutazindwa T, Patel AK, Somers K (1972) Survival after first presentation with endomyocardial fibrosis. Br Heart J 34: 403-407. [Crossref]
- Urhoghide GE, Falase AO (1987) Degranulated eosinophils, eosinophil granule basic proteins and humoral factors in Nigerians with endomyocardial fibrosis. Afr J Med Med Sci 16: 133-139. [Crossref]
- Fernández FJ, Berjón J, Ruiz V, Alcasena MS, Imizcoz MA, Lezáun R (2001) Tropical endomyocardial fibrosis or Davies disease: a case report. Rev Esp Cardiol 54: 235-238. [Crossref]
- Ellis J, Martin R, Wilde P, Tometzki A, Senkungu J, Nansera D (2007) Echocardiographic, chest X-ray and electrocardiogram findings in children presenting with heart failure to a Ugandan paediatric ward. Trop Doct 37: 149-150. [Crossref]
- Grimaldi A, Ammirati E, Karam N, Vermi AC, de Concilio A, et al. (2014) Cardiac surgery for patients with heart failure due to structural heart disease in Uganda: access to surgery and outcomes. Cardiovasc J Afr 25: 204-211. [Crossref]
- Verma VK, Zafar KS (2014) Tropical endomyocardial fibrosis: an overview. Int J Res Med Sci 2: 1267-1277.
- Goodwin JF (1983) Endomyocardial disease: clinical features. Postgrad Med J 59: 154-156. [Crossref]
- Blauwet LA, Breen JF, Edwards WD, Klarich KW (2005) Atypical presentation of eosinophilic endomyocardial disease. In Mayo Clinic Proceedings 80: 1078-1084. [Crossref]
- Seguela PE, Iriart X, Acar P, Montaudon M, Roudaut R, et al. (2015) Eosinophilic cardiac disease: molecular, clinical and imaging aspects. Arch Cardiovasc Dis 108: 258-268. [Crossref]
- Jaski BE, Goetzl EJ, Said JW, Fishbein MC (1978) Endomyocardial disease and eosinophilia. Report of a case. Circulation 57: 824-827. [Crossref]
- Brockington IF, Olsen EGJ (1973) Loeffler’s endocarditis and Davies’ endomyocardial fibrosis. Am Heart J 85: 308-322.
- Davies J, Spry CJ, Vijayaraghavan G, De Souza JA (1983) A comparison of the clinical and cardiological features of endomyocardial disease in temperate and tropical regions. Postgrad Med J 59: 179-185. [Crossref]
- Davies JN (1948) Endomyocardial fibrosis in Uganda. East Afr Med J 25: 10-16.
- Andy JJ (2001) Aetiology of endomyocardial fibrosis (EMF). West Afr J Med 20: 199-207. [Crossref]
- Andy JJ, Ogunowo PO, Akpan NA, Odigwe CO, Ekanem IA, Esin RA (1998) Helminth associated hypereosinophilia and tropical endomyocardial fibrosis (EMF) in Nigeria. Acta Trop 69: 127-140. [Crossref]
- Brockington IF, Olsen EG, Goodwin JF (1967) Endomyocardial fibrosis in Europeans resident in tropical Africa. Lancet 1: 583-588. [Crossref]
- Cilliers AM, Adams PE, Mocumbi AO (2011) Early presentation of endomyocardial fibrosis in a 22-month-old child: a case report. Cardiol Young 21: 101-103. [Crossref]
- Jaiyesimi F, Salimonu LS, Antia AU (1984) Serum immunoglobulins in children with cardiomyopathies. Trans R Soc Trop Med Hyg 78: 127-131. [Crossref]
- Mathai A, Kartha CC, Balakrishnan KG (1986) Serum immunoglobulins in patients with endomyocardial fibrosis. Indian Heart J 38: 470-472. [Crossref]
- Patel AK, Ziegler JL, D’Arbela PG, Somers K (1971) Familial cases of endomyocardial fibrosis in Uganda. Br Med J 4: 331-334. [Crossref]
- Van der Geld H, Peetoom F, Somers K, Kanyerezi BR (1966) Immunohistological and serological studies in endomyocardial fibrosis. Lancet 2: 1210-1213. [Crossref]
- Mocumbi AO, Latif N, Yacoub MH (2010) Presence of circulating anti-myosin antibodies in endomyocardial fibrosis. PLoS Negl Trop Dis 4: e661. [Crossref]
- Jaiyesimi F, Onadeko M, Antia AU (1979) Endomyocardial fibrosis, schistosomiasis and dermatosis: a new facet of an old problem? Tropical Cardiology 5: 27.
- Narain JP, Dash AP, Parnell B, Bhattacharya SK, Barua S, et al. (2010) Elimination of neglected tropical diseases in the South-East Asia region of the World Health Organization. Bull World Health Organ 88: 206-210. [Crossref]
- World Health Organization (2011) Weekly Epidemiological Record. Weekly Epidemiological Record 86: 389-400.
- Mayanja-Kizza H, Gerwing E, Rutakingirwa M, Mugerwa R, Freers J (2000) Tropical endomyocardial fibrosis in Uganda: the tribal and geographic distribution, and the association with eosinophilia. Trop Cardiol 103: 45-48.
- Shaper AG (1966) Endomyocardial fibrosis and rheumatic heart-disease. Lancet 1: 639-641. [Crossref]
- Shaper AG, Hutt MS, Coles RM (1968) Necropsy study of endomyocardial fibrosis and rheumatic heart disease in Uganda 1950-1965. Br Heart J 30: 391-401. [Crossref]
- Wayengera M (2009) Searching for new clues about the molecular cause of endomyocardial fibrosis by way of in silico proteomics and analytical chemistry. PLoS One 4: e7420. [Crossref]
- Sivasankaran S (2009) Restrictive cardiomyopathy in India: the story of a vanishing mystery. Heart 95: 9-14. [Crossref]
- Oke OL (1969) The role of hydrocyanic acid in nutrition. World Rev Nutr Diet 11: 170-198. [Crossref]
- Tylleskär T, Banea M, Bikangi N, Cooke RD, Poulter NH, Rosling H. Cassava cyanogens and konzo, an upper motoneuron disease found in Africa Lancet 339: 208-211 [Crossref]
- Nutman TB, Miller KD, Mulligan M, Ottesen EA (1986) Loa loa infection in temporary residents of endemic regions: recognition of a hyper-responsive syndrome with characteristic clinical manifestations. J Infect Dis 154: 10-18. [Crossref]
- Martin TN, Weir RA, Dargie HJ (2008) Contrast-enhanced magnetic resonance imaging of endomyocardial fibrosis secondary to Bancroftian filariasis. Heart 94: 1116. [Crossref]
- Beck W, Schrire V (1972) Endomyocardial fibrosis in Caucasians previously resident in tropical Africa. Br Heart J 34: 915-918. [Crossref]
- Chopra P, Narula J, Talwar KK, Kumar V, Bhatia ML (1990) Histomorphologic characteristics of endomyocardial fibrosis: an endomyocardial biopsy study. Hum Pathol 21: 613-616. [Crossref]
- Vazquez EF, Bautista CL, Navarrete BA, Moreno IC, Guerrero AE, et al. (2003) Chronic thromboembolic pulmonary hypertension associated with endomyocardial fibrosis of the right ventricle. Arch Bronconeumo 39: 370-372. [Crossref]
- Saraiva LR, Carneiro RW, Arruda MB, Brindeiro D, Lira V (1999) Mitral valve disease with rheumatic appearance in the presence of left ventricular endomyocardial fibrosis. Arq Bras Cardiol 72: 330-332. [Crossref]
- Ribeiro PA, Muthusami R, Duran CMG (1992) Rightsided endomyocardial fibrosis with recurrent pulmonary emboli leading to irreversible pulmonary hypertension. Br Heart J 68: 328-329. [Crossref]
- Jaiyesimi F (1982) Controversies and advances in endomyocardial fibrosis: a review. Afr J Med Med Sci 11: 37-46. [Crossref]
- Jaiyesimi F, Akinyemi OO, Falase AO (1977) Arterial oxygen desaturation in right endomyocardial fibrosis. Afr J Med Med Sci 6: 159-163. [Crossref]
- Rashwan MA, Ayman M, Ashour S, Hassanin MM, Zeina AA (1995) Endomyocardial fibrosis in Egypt: an illustrated review. Br Heart J 73: 284-289. [Crossref]
- Berensztein CS, Pinero G, Marcotegui M, Brunoldi R, Blanco MV, Lerman J (2000) Usefulness of echocardiography and Doppler echocardiographiy in endomyocardial fibrosis. J Am Soc Echocardiogr 13: 385-392. [Crossref]
- Mousseaux E, Hernigou A, Azencot M, Sapoval M, Auguste M, et al. (1996) Endomyocardial fibrosis: electron-beam CT features. Radiology 198: 755-760. [Crossref]
- Estornell J, Lopez MP, Dicenta F, Igual B, Martinez V, et al. (2003) Usefulness of magnetic resonance imaging in the assessment of endomyocardial disease. Rev Esp Cardiol 56: 321-324. [Crossref]
- Falase AO, Kolawole TM, Lagundoye SB (1976) Endomyocardial fibrosis: problems in differential diagnosis. Brit Heart J 38: 369-374. [Crossref]
- Chimenti C, Pieroni M, Frustaci A (2001) Endomyocardial fibrosis mimicking a dilated cardiomyopathy in a child. Heart 86: 73. [Crossref]
- Kleinfeldt T, Nienaber CA, Kische S, Akin I, Turan RG, et al. (2010) Cardiac manifestation of the hypereosinophilic syndrome: new insights. Clin Res Cardiol 99: 419-427. [Crossref]
- Graham JM, Lawrie GM, Feteih NM, Debakey ME (1981) Management of endomyocardial fibrosis: successful surgical treatment of biventricular involvement and consideration of the superiority of operative intervention. Am Heart J 102: 771-775. [Crossref]
- Moraes F, Lapa C, Hazin S, Tenorio E, Gomes C, et al. (1999) Surgery of endomyocardial fibrosis revisited. Eur J Cardio-Thoracic Surg 15: 30913. [Crossref]
- Valiathan MS, Balakrishnan KG, Sankarkumar R, Kartha CC (1987) Surgical treatment of endomyocardial fibrosis. Ann Thorac Surg 43: 68-73. [Crossref]
- Mocumbi AO, Sidi D, Vouhe P, Yacoub M (2007) An innovative technique for the relief of right ventricular trabecular cavity obliteration in endomyocardial fibrosis. J Thorac Cardiovasc Surg 134: 1070-1072. [Crossref]
- Joshi R, Abraham S, Kumar AS (2003) New approach for complete endocardiectomy in left ventricular endomyocardial fibrosis. J Thorac Cardiovasc Surg 125: 40-42. [Crossref]
- Yie K, Sung S, Kim D, Woo J (2004) Bidirectional cavopulmonary shunt as a rescue procedure for right ventricular endomyocardial fibrosis. Interact Cardiovasc Thorac Surg 3: 86-88. [Crossref]
- Anbarasu M, Krishna Manohar SR, Titus T, Neelakandhan KS (2004) One-an-a-half ventricle repair for right ventricular endomyocardial fibrosis. Asian Cardiovasc Thorac Ann 12: 363-365. [Crossref]
- Mishra A, Manohar SR, Ramalingam SK, Valiathan MS (2002) Bidirectional Glenn shunt for right ventricular endomyocardial fibrosis. Asian Cardiovasc Thorac An 10: 351-353. [Crossref]
- Da Costa FDA, Moraes CR, Rodrigues JV, de Mendonca JT, Andrade JC, et al. (1989) Early surgical results in the treatment of endomyocardial fibrosis. Eur J Cardio-Thorac Surg 3: 408-413. [Crossref]
- Moraes CR, Buffolo E, Moraes NF, Rodrigues JV, Gomes CA, et al. (1996) Recurrence of fibrosis after endomyocardial fibrosis surgery. Arq Bras Cardiol 67: 297-299. [Crossref]
- Freitas HFG, Castro PPN, Chizzola PR, Bocchi EA (2005) Transplante cardíaco em portadora de endomiocardiofibrose. Arq Bras Cardiol 84: 49-50.
- Chusid MJ, Dale DC, West BC, Wolff SM (1975) The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) 54: 1-27. [Crossref]
- Ogbogu PU, Bochner BS, Butterfield JH, et al. (2009) Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol 124: 1319-1325. [Crossref]
- Ommen SR, Seward JB, Tajik AJ (2000) Clinical and echocardiographic features of hypereosinophilic syndromes. Am J Cardiol 86: 110-113. [Crossref]
- Parrillo JE, Borer JS, Henry WL, Wolff SM, Fauci AS (1979) The cardiovascular manifestations of the hypereosinophilic syndrome. Prospective study of 26 patients, with review of the literature. Am J Med 67: 572-582. [Crossref]
- Spry CJ (1982) The hypereosinophilic syndrome: clinical features, laboratory findings and treatment. Allergy 37: 539-555. [Crossref]
- Loffler WL (1936) Endocarditis parietal fibroplastica with eosinophilia: a strange disease. Schweizerische Medizinische Wochenschrift 66: 817-820.
- Ogbogu PU, Rosing DR, Horne III MK (2007) Cardiovascular manifestations of hypereosinophilic syndromes. Immunol Allergy Clin North Am 27: 457-475. [Crossref]
- Bocquet H, Bagot M, Roujeau JC (1996) Drug-induced pseudolymphoma and drug hypersensitivity syndrome (drug rash with eosinophilia and systemic symptoms: DRESS). Semin Cutan Med Surg 15: 250-257. [Crossref]
- Boccara F, Benhaiem-Sigaux N, Cohen A (2001) Acute myopericarditis after diphtheria, tetanus, and polio vaccination. Chest 120: 671-672. [Crossref]
- Cassimatis DC, Atwood JE, Engler RM, Linz PE, Grabenstein JD, et al. (2004) Smallpox vaccination and myopericarditis: a clinical review. J Am Coll Cardiol 43: 1503-1510. [Crossref]
- Katz HT, Haque SJ, Hsieh FH (2005) Pediatric hypereosinophilic syndrome (HES) differs from adult HES. J Pediatr 146: 134-136. [Crossref]
- Fauci AS, Harley JB, Roberts WC, Ferrans VJ, Gralnick HR, et al. (1982) NIH conference. The idiopathic hypereosinophilic syndrome. Clinical, pathophysiologic, and therapeutic considerations. Ann Intern Med 97: 78-92. [Crossref]
- Weller PF, Bubley GJ (1994) The idiopathic hypereosinophilic syndrome. Blood 83: 2759-2779. [Crossref]
- Eicher JC, Bonnotte B, L’Huillier I, et al. (2009) Cardiovascular manifestations of eosinophilia: clinical and echocardiographic presentation. Rev Med Interne 30: 1011-1019. [Crossref]
- Ginsberg F, Parrillo JE (2005) Eosinophilic myocarditis. Heart Fail Clin 1: 419-429. [Crossref]
- Corradi D, Vaglio A, Maestri R, et al. (2004) Eosinophilic myocarditis in a patient with idiopathic hypereosinophilic syndrome: insights into mechanisms of myocardial cell death. Hum Pathol 35: 1160-1163. [Crossref]
- Krous HF, Haas E, Chadwick AE, Wagner GN (2005) Sudden death in a neonate with idiopathic eosinophilic endomyocarditis. Pediatr Dev Pathol 8: 587-592. [Crossref]
- D’Orazio J, Pulliam J (2011) Hypereosinophilic syndrome presenting as acute myocardial infarction in an adolescent. J Pediatr 158: 685. [Crossref]
- Amini R, Nielsen C (2010) Eosinophilic myocarditis mimicking acute coronary syndrome secondary to idiopathic hypereosinophilic syndrome: a case report. J Med Case Rep 4: 40. [Crossref]
- Chun W, Grist TM, Kamp TJ, Warner TF, Christian TF (2004) Images in cardiovascular medicine. Infiltrative eosinophilic myocarditis diagnosed and localized by cardiac magnetic resonance imaging. Circulation 110: e19. [Crossref]
- Aslan I, Fischer M, Laser KT, Haas NA (2013) Eosinophilic myocarditis in an adolescent: a case report and review of the literature. Cardiol Young 23: 277-283. [Crossref]
- May LJ, Patton DJ, Fruitman DS (2011) The evolving approach to paediatric myocarditis: a review of the current literature. Cardiol Young 21: 241-251. [Crossref]
- Yanagisawa T, Inomata T, Watanabe I, et al. (2011) Clinical significance of corticosteroid therapy for eosinophilic myocarditis. Int Heart J 52: 110-113. [Crossref]
- Slungaard A, Vercellotti GM, Tran T, Gleich GJ, Key NS (1993) Eosinophil cationic granule proteins impair thrombomodulin function. A potential mechanism for thromboembolism in hypereosinophilic heart disease. J Clin Invest 91: 1721-1730. [Crossref]
- Zientek DM, King DL, Dewan SJ, Harford PH, Youman DJ, et al. (1995) Hypereosinophilic syndrome with rapid progression of cardiac involvement and early echocardiographic abnormalities. Am Heart J 130: 1295-1298. [Crossref]
- Hoffman M, Monroe DM III (2001) A cell-based model of haemostasis. Thromb Haemost 85: 958-965. [Crossref]
- Ruggeri ZM (2006) Platelet interactions with vessel wall components during thrombogenesis. Blood Cells Mol Dis 36: 145-147. [Crossref]
- Wang JG, Mahmud SA, Thompson JA, Geng JG, Key NS, et al. (2006) The principal eosinophil peroxidase product, HOSCN, is a uniquely potent phagocyte oxidant inducer of endothelial cell tissue factor activity: a potential mechanism for thrombosis in eosinophilic inflammatory states. Blood 107: 558-565. [Crossref]
- Moosbauer C, Morgenstern E, Cuvelier SL, et al. (2007) Eosinophils are a major intravascular location for tissue factor storage and exposure. Blood 109: 995-1002. [Crossref]
- Venge P, Dahl R, Hallgren R (1979) Enhancement of factor XII dependent reactions by eosinophil cationic protein. Thromb Res 14: 641-649. [Crossref]
- Gambacorti Passerini C, Cortellaro M, Cofrancesco E, et al. (1989) Possible mechanisms of fibrin deposition in the hypereosinophilic syndrome. Haemostasis 19: 32-37. [Crossref]
- Rohrbach MS, Wheatley CL, Slifman NR, Gleich GJ (1990) Activation of platelets by eosinophil granule proteins. J Exp Med 172: 1271-1274. [Crossref]
- Spry CJ, Davies J, Ta i PC, Olsen EG, Oakley CM, Goodwin JF (1983) Clinical features of fifteen patients with the hypereosinophilic syndrome. Q J Med 52: 1-22. [Crossref]
- Korczyk D, Taylor G, McAlistair H, et al. (2007) Heart transplantation in a patient with endomyocardial fibrosis due to hypereosinophilic syndrome. Transplantation 83: 514-516. [Crossref]
- Carnero-Alcazar M, Reguillo-Lacruz F, O’Connor F, Rodriguez-Hernandez E (2008) Hypereosinophilic syndrome and myocardial fibrosis. Interact Cardiovasc Thorac Surg 7: 928-930. [Crossref]
- Fuzellier JF, Chapoutot L, Torossian PF (2004) Mitral valve repair in idiopathic hypereosinophilic syndrome. J Heart Valve Dis 13: 529-531. [Crossref]
- Salanitri GC (2005) Endomyocardial fibrosis and intracardiac thrombus occurring in idiopathic hypereosinophilic syndrome. AJR Am J Roentgenol 184: 1432-1433. [Crossref]
- Hendren WG, Jones EL, Smith MD (1988) Aortic and mitral valve replacement in idiopathic hypereosinophilic syndrome. Ann Thorac Surg 46: 570-571. [Crossref]
- Gottdiener JS, Maron BJ, Schooley RT, Harley JB, Roberts WC, et al. (1983) Two-dimensional echocardiographic assessment of the idiopathic hypereosinophilic syndrome. Anatomic basis of mitral regurgitation and peripheral embolization. Circulation 67: 572-578. [Crossref]
- Dedieu N, Giardini A, Khambadkone S, Marek J (2011) Eosinophilic heart disease in a paediatric patient. Eur J Echocardiogr 12: E3. [Crossref]
- Assayag P, Carre F, Chevalier B, Delcayre C, Mansier P, et al. (1997) Compensated cardiac hypertrophy: arrhythmogenicity and the new myocardial phenotype. I. Fibrosis. Cardiovasc Res 34: 439-444. [Crossref]
- Spiegel R, Miron D, Fink D, Gavriel H, Horovitz Y (2004) Eosinophilic pericarditis: a rare complication of idiopathic hypereosinophilic syndrome in a child. Pediatr Cardiol 25: 690-692. [Crossref]
- D’Souza MG, Swistel DG, Castro JL, DeRose JJ Jr (2003) Hypereosinophilic thrombus causing aortic stenosis and myocardial infarction. Ann Thorac Surg 7: 1725-1726. [Crossref]
- Cools J, DeAngelo DJ, Gotlib J, et al. (2003) A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 348: 1201-1214. [Crossref]
- Subhash HS, Asishkumar M, Jonathan M (2002) Unusual cardiac manifestation of hypereosinophilic syndrome. Postgrad Med J 78: 490-491. [Crossref]
- Shah R, Ananthasubramaniam K (2006) Evaluation of cardiac involvement in hypereosinophilic syndrome: complementary roles of transthoracic, transesophageal, and contrast echocardiography. Echocardiography 23: 689-691. [Crossref]
- Syed IS, Martinez MW, Feng DL, Glockner JF (2008) Cardiac magnetic resonance imaging of eosinophilic endomyocardial disease. Int J Cardiol 126: e50-52. [Crossref]
- Wagner A, Mahrholdt H, Holly TA, et al. (2003) Contrast-enhanced MRI and routine single photon emission computed tomography (SPECT) perfusion imaging for detection of subendocardial myocardial infarcts: an imaging study. Lancet 361: 374-379. [Crossref]
- Lofiego C, Ferlito M, Rocchi G, Biagini E, Perugini E, et al. (2005) Ventricular remodeling in Loeffler endocarditis: implications for therapeutic decision making. Eur J Heart Fail 7: 1023-1026. [Crossref]
- Rothenberg ME, Klion AD, Roufosse FE, Kahn JE, Weller PF, et al. (2008) Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med 358: 1215-1228. [Crossref]
- Legrand F, Klion AD (2015) Biologic therapies targeting eosinophils: current status and future prospects. J Allergy Clin Immunol Pract 3: 167-174. [Crossref]
- Weinberg T, Himelfarb AJ (1943) Endocardial fibroelastosis (so-called fetal endocarditis). A report of two cases occurring in siblings. Bull Johns Hopkins Hosp 72: 299-306.
- David EP, Arévalo RP, Fernández-Avilés F (2008) Endocardial fibroelastosis in dilated cardiomyopathy in a 28-year-old transplant recipient. Eur Heart J 30: 477. [Crossref]
- Hunter AS, Keay AJ (1973) Primary endocardial fibroelastosis. An inherited condition. Arch Dis Child 48: 66-69. [Crossref]
- Seki A, Patel S, Ashraf S, Perens G, Fishbein MC (2013) Primary endocardial fibroelastosis: an underappreciated cause of cardiomyopathy in children. Cardiovasc Pathol 22: 345-350. [Crossref]
- Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, et al. (1997) Epidemiology of idiopathic cardiomyopathies in children and adolescents: a nationwide study in Finland. Am J Epidemiol 146: 385-393. [Crossref]
- Arya SO, Karpawich PP, Gupta P, Buddhe S, Singh HR, et al. (2012) Primary endocardial fibroelastosis presenting in a young child as incessant ventricular tachycardia and dilated cardiomyopathy. Tex Heart Inst J 39: 714-718. [Crossref]
- Ni J, Bowles NE, Kim YH, et al. (1997) Viral infection of the myocardium in endocardial fibroelastosis. Molecular evidence for the role of mumps virus as an etiologic agent. Circulation 95: 133-139. [Crossref]
- Lloyd DF, Vara R, Mathur S (2017) Cardiac manifestations of inherited metabolic disease in children. Pediatrics International 59: 525-529. [Crossref]
- Gilbert-Barness E (2004) Metabolic cardiomyopathy and conduction system defects in children. Ann Clin Lab Sci 34: 15-34. [Crossref]
- Nield LE, Silverman ED, Taylor GP, Smallhorn JF, Mullen JB, et al. (2002) Maternal anti-Ro and anti-La antibody–associated endocardial fibroelastosis. Circulation 105: 843-848. [Crossref]
- Nield LE, Smallhorn JF, Benson LN, Hornberger LK, Silverman ED, et al. (2002) Endocardial fibroelastosis associated with maternal anti-Ro and anti-La antibodies in the absence of atrioventricular block. J Am Coll Cardiol 40: 796-802. [Crossref]
- Chen S, Thompson MW, Rose V (1971) Endocardial fibroelastosis: family studies with special reference to counseling. J Pediatr 79: 385-392. [Crossref]
- Fan Y, Steller J, Gonzalez IL, Kulik W, Fox M, et al. (2013) A Novel Exonic Splicing Mutation in the TAZ (G4. 5) Gene in a case with atypical Barth syndrome. JIMD Rep 11: 99-106. [Crossref]
- Steward CG, Newbury‐Ecob RA, Hastings R, Smithson SF, Tsai‐Goodman B, et al. (2010) Barth syndrome: an X‐linked cause of foetal cardiomyopathy and stillbirth. Prenat Diagn 30: 970-976. [Crossref]
- Sinha SK, Aggarwal P, Rekwal L, Tripathi S, Abhishekh NK (2018) Unexplained heart failure in a 14-month baby. Folia Cardiologica 13: 556-560.
- Moller JH, Lucas RV, Adams P, et al. (1964) Endocardial fibroelastosis: a clinical and anatomic study of 47 patients with emphasis on its relationship to mitral insufficiency. Circulation 30: 759-782. [Crossref]
- Keith JD, Rose V, Manning JA. Endocardial fibroelastosis. In Keith JD, Rowe RD, Vlad P, eds. Heart Disease in Infancy and Childhood. 3rd ed. New York, NY: MacMillan; 1978. 941-957.
- Sellers FJ, Keith JD, Manning JA (1964) The diagnosis of primary endocardial fibroelastosis. Circulation 29: 49-59. [Crossref]
- Maredia N, English K, Greenwood J (2008) Assessment of endocardial fibroelastosis by cardiac MRI. Can J Cardiol 24: e33. [Crossref]
- Stranzinger E, Ensing GJ, Hernandez RJ (2008) MR findings of endocardial fibroelastosis in children. Pediatr Radiol 38: 292-296. [Crossref]
- Zhorne D, Petit CJ, Ing FF, Justino H, Jefferies JL, Dreyer WJ (2013) A 25-year experience of endomyocardial biopsy safety in infants. Catheter Cardiovasc Interv 82: 797-801. [Crossref]
- Ozdemir D, Cortopassi IO, McNamara RL (2019) An illustrative case of endocardial fibroelastosis and recalcitrant intracardiac thrombosis: a case report. Thrombosis Journal 17: 8. [Crossref]
- Costedoat-Chalumeau N, Amoura Z, Villain E, Cohen L, Piette JC (2005) Anti-SSA/Ro antibodies and the heart: more than complete congenital heart block? A review of electrocardiographic and myocardial abnormalities and of treatment options. Arthritis Res Ther 7: 69. [Crossref]
- Steger CM, Antretter H, Moser PL (2012) Endocardial fibroelastosis of the heart. Lancet 379: 932. [Crossref]
- Schmaltz AA, Ibrahim Z, Heil RP, Brown DJ (1979) Echocardiographic Findings and Function Analysis in Infants with Endocardial Fibroelastosis. Echocardiology 345-348. [Crossref]
- Dogan V, Ozgur S, Ertugrul I, Yoldas T, Orun UA, et al. (2015) Clinical characteristics and outcomes in patients with endocardial fibroelastosis associated with critical aortic stenosis with biventricular circulation. Turkish Journal of Thoracic and Cardiovascular Surgery.