Tricho-hepato-enteric syndrome is a rare autosomal recessive enteropathy that first presents as intractable diarrhea in neonates. Diarrhea persists throughout life and patients are dependent on parenteral nutrition for growth. Additional features include facial dysmorphism, trichorrhexis nodosa (woolly hair), intra-uterine growth restriction, hepatic disease, skin anomalies and a depressed immune system. Tricho-hepato-enteric syndrome is a life limiting disease with variability in its manifestations and severity. Mutations in two different genes, TTC37 or SKIV2L, cause the disorder.
In this case report we present a neonate with a novel mutation in TTC37 that resulted in a severe phenotype associated with fulminate sepsis. The infant presented at one week of age with sudden onset of diarrhea and dehydration. Tricho-hepato-enteric syndrome was diagnosed by whole exome sequencing but was not initially considered because the infant lacked many of the diagnostic clinical features. Soon after presentation, the infant developed pneumoperitoneum and necrosis of entire bowel. The blood culture was positive for Clostridium perfringens. Autopsy showed bacteria in the parenchyma and vasculature of all major internal organs as well as within the bone marrow, connective tissue and skeletal muscle but there was minimal inflammatory response. The lack of migration of white blood cells to the sites of infection is likely due to the combined immunodeficiency reported in patients with tricho-hepato-enteric syndrome.
This case expands our knowledge on the clinical features of tricho-hepato-enteric syndrome. Whole-exome sequencing was instrumental in making the diagnosis in an infant with an atypical presentation and should be considered early in neonates with congenital diarrhea.
congenital diarrhea, Clostridium perfringens, enterocolitis, tricho-hepato-enteric syndrome
THES: Tricho-hepato-enteric syndrome; TTC37: Tetratricopeptide repeat domain 37; SKIV2L: Ski2 like RNA helicase; SKI: Superkiller tetraprotein; TRP: Tetratricopeptide; NEC: Necrotizing enterocolitis
Congenital diarrhea disorders encompass a group of rare inherited intestinal diseases that present during the first months of life with intractable diarrhea leading to malabsorption and failure to thrive. There are various etiologies of congenital diarrhea including those due to defective enterocytes. One such disorder is tricho-hepato-enteric syndrome (THES) also known as syndromic diarrhea, an autosomal recessive neonatal enteropathy with an incidence between 1/400,000 and 1/1,000,000 [1,2]. THES is due to a mutation in either of two genes Tetratricopeptide Repeat Domain 37 (TTC37) and Ski2 Like RNA Helicase (SKIV2L) [3-5]. Both TTC37 and SKIV2L encode components of the SKI (superkiller) tetraprotein which binds to cytoplasmic exosomes [1] and facilitate mRNA processing and degradation [6]. Specific to the mutation observed in our patient, TTC37 encodes the protein Thespin which is comprised of 20 tetratricopeptide (TRP) repeated domains [3,4]. TRP repeats mediate protein-protein interactions and the assembly of multiprotein complexes. The scaffolding of the SKI tetraprotein complex is supported by the TRP repeats within Thespin [7]. Pathologic mutations occur throughout the gene without a mutation hot spot and include splicing abnormalities, frameshifts induced by deletions or insertions and missense mutations [1].
Nearly 100% of patients with THES have intractable diarrhea that persists despite enteral rest [1,8,9]. In a review of 15 patients with THES, the onset of diarrhea began at a median age of 24 days with a range of 0-335 days. Stools are watery with a 25% incidence of hematochezia. There is normal intestinal mucosa in 25%, mild villous atrophy in 50% and subtotal villous atrophy in 25% of patients [9]. Diarrhea continues throughout life and is managed with supplemental parenteral nutrition [8]. The probability of totally weaning off parenteral nutrition is 0%, 10% and 40% at 1, 5 and 10 years respectively [9]. In addition to congenital diarrhea, the majority of infants with THES have facial dysmorphism, wooly hair from trichorrhexis nodosa, intra-uterine growth restriction, and abnormalities of the immune system. The most common facial features include a wide forehead, broad nasal root, hypertelorism and a course facial appearance [5]. Although “hepato” is a component of the name, only 50% of patients with THES have liver anomalies which include elevated liver enzymes, cirrhosis and hemochromatosis [10]. Hepatic involvement is inconsistent even among siblings with the same mutation. TTC37 is widely expressed in vascular tissues, lymph node, pituitary, lung, and intestine but is not expressed in the liver [3]. However, the hepatic manifestation of THES may begin before the onset of diarrhea, so are not entirely due to the detrimental effects of parenteral nutrition [5]. Skin abnormalities such xerosis, café-au-lait spots and angioma are also present in about half of the patients [9]. Congenital heart defects and platelet abnormalities are occasionally reported. Mild intellectual disabilities are present in 50% of patients which may be inherent to THES or a consequence of prolonged nutritional deficiency and postanal growth failure. Magnetic resonance imaging of the brain is usually normal [2].
It is unknown how mutations in TTC37 with the consequent alteration in processing and degradation of RNA precisely lead to all the manifestations of THES. There are no specific diagnostic features of THES on histologic examination of intestinal biopsy specimens. The morphology of the brush border is usually normal; however, immunohistochemical studies demonstrate that the enzymes and transporter proteins typically associated with the apical brush border are aberrantly located within enterocytes [5,11]. This would indicate a generalized defect in transport of intracellular proteins. A review of all reported mutations in TTC37 and SKIV2L which included 96 patients from 18 different families was published in 2018. Only two recurrent mutation have been identified in independent families. There is minimal correlation between the genotype and phenotype, mostly likely due to the broad spectrum of mutations [1,12]. This case introduces a novel mutation that when expressed with another rare mutation of TTC37 results in a severe phenotype associated with total bowel necrosis, fulminant sepsis and early death.
A 7-day old African American female presented to the emergency room with a one-day history of diarrhea and weight loss. She was born at 39 weeks’ gestation by a scheduled cesarean delivery. There was no history of consanguinity or complications with the pregnancy. The infant has two full siblings who have no chronic medical conditions. Prenatal laboratory screens were negative other than group B streptococcus unknown. Because the amniotic membranes were artificially ruptured at the time of delivery, no prophylactic antibiotics were administered. The Apgar scores were 8 and 9 at 1 and 5 minutes of age respectfully. The infant’s birth weight was 3.38 kg (50%) and length was 51.5 cm (80%). She had an uncomplicated postnatal course and was discharge from the hospital at two days of age. Her initial visit to the pediatrician was at five days of age at which time she was 2% below her birth weight and was orally feeding 1.5-2 ounces of formula every two hours. Shortly after the initial pediatric visit, the infant developed tan watery stools necessitating approximately 20 diaper changes within 24 hours. She was seen by her pediatrician the following day and by that time had lost 12% of her birth weight and had a hemoccult positive stool. No household contacts had diarrhea but both the infant’s siblings had upper respiratory infections. There was a positive family history for milk protein allergy in the infant’s cousin and one of her siblings. The pediatrician instructed the family to bring the infant to the emergency room for rehydration.
In the emergency room the infant appeared jaundice with dry mucus membranes, a sunken anterior fontanelle, and delayed capillary refill. She was irritable but had a normal abdominal exam and continued to take formula from a bottle. She was afebrile with normal vital signs other than tachycardia. No dysmorphology or skin anomalies were identified. Peripheral intravenous access was established and she was rehydrated with saline boluses followed by a D5W ½ normal saline infusion. The initial serum chemistries showed mild metabolic acidosis with minimal hyponatremia, hyperkalemia and an elevated serum creatinine. Her peak total serum bilirubin concentration was 19.0 mg/dL which decreased with rehydration; direct bilirubin was 0.7 mg/dL. Her complete blood count was normal with a white blood cell count of 10,000/cumm and platelet count of 234,000/cumm. There were no immature neutrophils and the absolute neutrophil and lymphocyte counts were within the 5-95% for a healthy newborn. Her c-reactive protein was 1.0 mg/dl. The urine culture was negative and the stool culture was negative for Salmonella, Shigella, Campylobacter and Escherichia coli 0157. The stool was also negative for Shigella toxin and the enzyme immunoassay for rotavirus was negative. Her initial clotting studies were normal and liver enzymes were mildly elevated. During her short hospital course, the liver enzymes increased to the following concentrations: alanine aminotransferase 328 U/L (normal 7–52 U/L), aspartate aminotransferase 139 U/L (normal 13–39 U/L), and alkaline phosphatase 331 U/L (normal 88–351 U/L). The peak concentration of ammonia was 399 mCmol/L which spontaneously decreased to 198 mCmol/L (normal 6-47 mCmol/L). A hepatitis panel was negative and cultures for cytomegalovirus and herpes simplex virus were also negative.
Because of difficulty maintaining peripheral intravenous access, she was transferred to the newborn intensive care unit. Enteral nutrition was discontinued and an umbilical venous catheter was placed for continued intravenous rehydration. Approximately 12 hours after admission to the hospital, she passed a copious amount of bright red blood per rectum. At the time her blood culture was pending, however broad-spectrum antibiotics were started after she developed rectal bleeding. Her abdominal exam continued to be normal with no tenderness or distension. An abdominal radiograph and ultrasound were normal without distended loops of bowel, ascites or evidence of pneumatosis intestinalis. Pediatric surgery and gastroenterology were consulted. A flexible sigmoidoscopy was performed which showed the rectosigmoid colon was inflamed, edematous and friable. Surgical pathology reported necrotic and devitalized soft tissue with bacterial colonization.
Shortly after the sigmoidoscopy, the infant clinically deteriorated with a worsening metabolic acidosis and hyperkalemia. She developed pneumoperitoneum and an emergent exploratory laparotomy was performed at the bedside. There was extensive intestinal necrosis from the jejunum to the sigmoid colon. The entire colon and all but 30 cm of small intestine proximal to the ligament of Treitz were resected. She had an unstable post-operative course and developed bradycardia, hypotension and poor perfusion that did not respond to resuscitative efforts. The infant expired approximately 24 hours after admission to the hospital. An autopsy was performed and a buccal sample was collected for exome sequence analysis. Posthumously, the blood culture obtained on admission to the hospital was reported positive for Clostridium perfringens.
Follow-up results
The autopsy demonstrated cysts filled with gram-positive rods on the surfaces of the liver (Figure 1a), spleen, kidneys and adrenal glands. Gram-positive rods were present in the parenchyma and vasculature of all major internal organs as well as within the bone marrow, connective tissue and skeletal muscle. The hepatic sinusoids were dilated with gram-positive rods (Figure 1b) and there was patchy hepatocyte necrosis but no fibrosis. The spleen, and thymus were normal in size with necrosis and gram-positive rods present. Lymph nodes had dilated sinusoids with gram-positive rods within vessels. Cultures of the lung, spleen and blood grew Clostridium perfringens. Despite evidence of overwhelming sepsis, there was a striking paucity of inflammatory cells in the parenchyma and vasculature of all the organs with abundant bacteria. No congenital anomalies or dysmorphic features were identified.
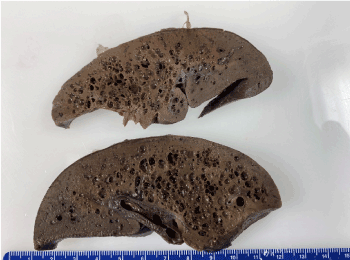
Figure 1. The liver was slightly over the expected weight with numerous cysts on the surface and throughout the parenchyma
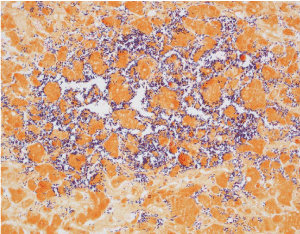
Figure 2. Gram stain of the liver at 40x magnification. There are abundant gram positive, rod-shaped bacteria within dilated hepatic sinusoids with no inflammatory reaction
The exome sequence analysis (GeneDx®) showed the infant was heterozygous for two variants of the TTC37 gene.
p.Leu1195Val (CTT>GTT): c.3583 C>G in exon 34
p.Glu1314Lys (GAA>AAA): c3940 G>A in exon 38
The variants are described according to the most recent mutation nomenclature guidelines [13]. Both mutations are missense variants that have not been reported as either pathologic or benign. p.Glu1314Lys (GAA>AAA): c3940 G>A in exon 38 has been found in 0.092% of large population cohorts but p.Leu1195Val (CTT>GTT): c.3583 C>G in exon 34 has not been previously observed [14]. The parents did not undergo genetic testing; therefore, it was not confirmed that the two variants of TTC37 were on separate alleles.
Discussion and conclusion
When the infant initially presented to the hospital the differential diagnosis for the diarrhea included infectious causes, glucose-galactose malabsorption and inherited diarrheas such as microvillous inclusion disease, tufting enteropathy, congenital chloridorrhea and congenital sodium diarrhea [11]. With the bleeding per rectum, the differential diagnosis expanded to include milk protein allergy, infantile polyp, anal fissure, Meckel's diverticulum, intussusception and necrotizing enterocolitis (NEC). Due to the lack of several specific features, THES was not considered in the initial differential diagnosis. Approximately 80-90% of infants with THES have in utero growth restriction [1]. The infant in this case was appropriately grown for her gestational age. No dysmorphic features, hair or skin anomalies were found on physical exam; however, facial dysmorphism, hair and skin anomalies may not be readily apparent in the newborn period. The elevated ammonia, liver enzymes and hepatocyte necrosis found on autopsy may have been a manifestation of THES or from her overwhelming sepsis. Iron stains of the liver to evaluate for hemochromatosis were not performed but if positive would have only been a marker for severe neonatal liver disease [15] and not necessarily indicative of THES. Since no genetic evaluation was performed on the parents, the diagnosis of THES in this infant is not completely certain because the disease is autosomal recessive and we were not able to confirm the mutations are in the trans position. However, in an update on 65 patients with phenotypic characteristics of THES and mutations in TTC37, 8% had the mutation on only 1 allele [1]. The authors concluded that in these cases, the second mutation maybe within an intron or another gene that affects regulation of TTC37.
The infant’s benign abdominal exam, normal complete blood count, c-reactive protein and initial radiographic studies were not consistent with the diagnosis of enterocolitis with necrosis of the entire bowel. Infants born at greater than 30 weeks’ gestation who develop NEC usually have an associated risk factor such as congenital heart disease, hypoxia, intestinal malformations or maternal drug use [16]. The infant in this case had no clear predisposing risk factor or indication of bowel necrosis when she first presented to the hospital. However, disruption of normal colonization of the intestinal track is a risk factor for NEC preterm infants [17]. This infant was at increased risk for the development of intestinal dysbiosis because she was born by cesarean delivery. Compared to vaginally-born infants, infants born by cesarean section have a higher carriage rate of Clostridium perfringens and fewer protective facultative anaerobes in their stool during the first 6 months [18]. In addition, she was primarily fed formula which does not have the commensal protective bacteria found in breast milk [19]. In this case, the intestinal microbiota was likely colonized with Clostridium perfringens. Enterocolitis associated with Clostridium perfringens has an earlier onset and a more fulminate course with increased necrosis and mortality compared to enterocolitis associated with other organisms or no causative agent [20] The majority of infants with THES have normal morphology of the brush border with mild to no villous atrophy [5,9,11]. However, the unique genetic defect in this infant likely lead to a breakdown of intestinal integrity. Colonization with an enteric pathogen along with the breakdown of intestinal integrity resulted in sepsis and early death in this patient.
The autopsy revealed an almost complete absence of an inflammatory response to invasive bacteria. This may be due to how quickly the infant succumbed to the infection. Alternatively, it may be secondary to the immunodeficiency associated with THES. Patients with THES have low serum concentrations of immunoglobulins and a poor immunologic response to childhood vaccination [5]. Although most patients with THES are managed with infusions of immunoglobin, 20% still ultimately die from infection [9]. An immunologic evaluation of a cohort of patients with THES found decreased memory B cells, abnormal T-cell proliferation, reduced degranulation of NK cells and decreased IFN-ɣ production from T-cells and NK cells [12].
Whole exome sequencing was invaluable in making the diagnosis and counseling the family. The etiology of congenital diarrhea is diverse and includes anomalies of enterocytes that are not always distinguishable by biopsy [11], especially with superimposed enterocolitis. Without exome sequencing, the infant would have been misdiagnosed as simply an atypical presentation of NEC. Because two mutations of TTC37 were identified, the family has the option of prenatal testing in the future. Survival in patients with THES is reported to be 83.3%, 63.4%, 54.9% and 50.7%, at 1, 5, 10 and 15 years respectively [8]. Although THES is a life limiting disease, previously reported patients do not die as young as the patient presented here. For example, mutations affecting the C-terminal end of the protein cause a mild phenotype with better growth and less dependence on parenteral nutrition [21]. The mutations described here were not associated with in utero growth restriction but resulted in a severe phenotype complicated by enterocolitis and necrosis of the entire bowel, sepsis and early death. These mutations should be added to the database of TTC37 mutations potentially causing a severe phenotype that puts infants at increased risk of developing fulminant sepsis and death. This case expands our knowledge of the varied clinical manifestations of THES and supports its inclusion in the differential diagnosis of congenital diarrhea in newborns without the typical phenotypic features previously reported.
- Patrice B, Clothilde E, Charlène C, Christophe B, Nicolas L, et al. (2018) Tricho-hepato-enteric syndrome mutation update: Mutations spectrum of TTC37 and SKIV2L, clinical analysis and future prospects. Hum Mutat 39: 774-789. [Crossref]
- Fabre A, Bourgeois P, Chaix C, Bertaux K, Goulet O, et al. (1993) Trichohepatoenteric syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K & Amemiya A. (eds.) GeneReviews((R)). Seattle (WA).
- Fabre A, Martinez-Vinson C, Roquelaure B, Missirian C, André N, et al. (2011) Novel mutations in TTC37 associated with tricho-hepato-enteric syndrome. Hum Mutat 32: 277-281. [Crossref]
- Kinnear C, Glanzmann B, Banda E, Schlechter N, Durrheim G, et al. (2017) Exome sequencing identifies a novel TTC37 mutation in the first reported case of trichohepatoenteric syndrome (THE-S) in South Africa. BMC Med Genet 18: 26.
- Hartley JL, Zachos NC, Dawood B, Donowitz M, Forman J, et al. (2010) Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy). Gastroenterology, 138: 2388-98, 2398 e1-2. [Crossref]
- Morton DJ, Kuiper EG, Jones SK, Leung SW, Corbett AH, et al. (2018) The RNA exosome and RNA exosome-linked disease. RNA, 24: 127-142. [Crossref]
- Zeytuni N, Zarivach R (2012) Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure 20: 397-405. [Crossref]
- Fabre A, Martinez-Vinson C, Goulet O, Badens C (2013) Syndromic diarrhea/Tricho-hepato-enteric syndrome. Orphanet J Rare Dis 8: 5.
- Fabre A, Breton A, Coste ME, Colomb V, Dubern B, et al. (2014) Syndromic (phenotypic) diarrhoea of infancy/tricho-hepato-enteric syndrome. Arch Dis Child 99: 35-38. [Crossref]
- Dweikat I, Sultan M, Maraqa N, Hindi T, Abu-Rmeileh S, et al. (2007) Tricho-hepato-enteric syndrome: A case of hemochromatosis with intractable diarrhea, dysmorphic features, and hair abnormality. Am J Med Genet A 143A: 581-583. [Crossref]
- Overeem AW, Posovszky C, Rings EH, Giepmans BN, Van ISC (2016) The role of enterocyte defects in the pathogenesis of congenital diarrheal disorders. Dis Model Mech 9: 1-12. [Crossref]
- Vely F, Barlogis V, Marinier E, Coste ME, Dubern B, et al. (2018) Combined immunodeficiency in patients with trichohepatoenteric syndrome. Front Immunol 9: 1036. [Crossref]
- Den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, et al. (2016) HGVS recommendations for the description of sequence variants: 2016 Update. Hum Mutat 37 564-569. [Crossref]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285-291. [Crossref]
- Whitington PF (2012) Gestational alloimmune liver disease and neonatal hemochromatosis. Semin Liver Dis 32: 325-32. [Crossref]
- Abbo O, Harper L, Michel JL, Ramful D, Breden A, et al. (2013) Necrotizing enterocolitis in full term neonates: is there always an underlying cause? J Neonatal Surg 2: 29. [Crossref]
- Frost BL, Modi BP, Jaksic T, Caplan MS (2017) New medical and surgical insights into neonatal necrotizing enterocolitis: A Review. JAMA Pediatr 171: 83-88. [Crossref]
- Nagpal R, Tsuji H, Takahashi T, Nomoto K, Kawashima K, et al. (2017) Gut dysbiosis following C-section instigates higher colonisation of toxigenic Clostridium perfringens in infants. Benef Microbes 8: 353-365. [Crossref]
- Sullivan S, Schanler RJ, Kim JH, Patel AL, Trawoger R, et al. (2010) An exclusively human milk-based diet is associated with a lower rate of necrotizing enterocolitis than a diet of human milk and bovine milk-based products. J Pediatr 156: 562-567 e1. [Crossref]
- Dittmar E, Beyer P, Fischer D, Schafer V, Schoepe H, et al. (2008) Necrotizing enterocolitis of the neonate with Clostridium perfringens: Diagnosis, clinical course, and role of alpha toxin. Eur J Pediatr 167: 891-895. [Crossref]
- Fabre A, Petit LM, Hansen LF, Wewer AV, Esteve C, et al. (2018) A new mutation in the C-terminal end of TTC37 leading to a mild form of syndromic diarrhea/tricho-hepato-enteric syndrome in seven patients from two families. Am J Med Genet A 176: 727-732. [Crossref]