Episodic ataxia is a heterogeneous group of disorders with an autosomal dominant pattern of inheritance mostly; however sporadic cases are also reported. It is characterized by attacks of incoordination, imbalance and progressive truncal ataxia that may last for minutes to few hours.
This case report describes a 45 years old Omani female, known case of chronic migraine headaches since childhood and with positive family history of migraine headache, presenting with attacks of vertigo, ataxia and interictal nystagmus. These attacks increased significantly, in frequency and duration, without obvious precipitating factor. The patient was diagnosed clinically to have episodic ataxia and proved by a positive genetic testing showing heterogeneity for missense mutation in the CACNB4 gene with c.1412G>A (p.Arg471Lys) variant. Her episodes improved successfully to treatment with Acetazolamide.
episodic ataxia, EA5, CACNB4, acetazolamide
Episodic ataxia (EA) is a rare autosomal dominant inherited neurological disorder characterized by bouts of incoordination and progressive ataxia [1,2]. It’s a heterogeneous condition, clinically and genetically [3]. Clinically, EAs are heterogeneous and the ataxic spells are variably associated with vertigo, tinnitus, diplopia, blurred vision, myokymia, tremor, obtundation, or headache and may coexist with epilepsy and alternating hemiplegia [4-6]. The attacks can happen spontaneously and can be triggered by certain factors, like alcohol or coffee intake, lack of sleep, exercise, fatigue, or anxiety. Permanent ataxia and cerebellar atrophy may develop after years of disease duration [2,7].
Genetically, eight sub-types of EA are identified, till now [6] and the majority of cases reported are of subtypes EA-type 1 (EA1) and EA-type 2 (EA2) which are caused by mutations in the potassium and calcium channel genes KCNA1 and CACNA1A, respectively. Both genetically confirmed EA1 and EA2 cases have been demonstrated in multiple families [1,7] mostly of Caucasian ethnicity [2]. EA-type 5 (EA5) and EA-type 6 (EA6) are known to be caused by mutations in CACNB4 and SLC1A3, respectively, but have been reported in only one or two families [8-10]. In EA4, known EA genes were excluded and distinct disease loci were mapped for EA3 and EA7 [11-13]. A recent reported study, which used whole-exome sequencing (WES), revealed an association of UBR4 with EA8 [14].
Migraine association with episodic ataxia is reported in types EA1 and EA2 patients and it is also well known to be associated with periodic vestibule-cerebellar ataxia and with episodic ataxia with paroxysmal choreoathetosis and spasticity [15-17]. On the other hand, episodic ataxia is seen in basilar type migraine; as well as in familial hemiplegic migraine [15].
The aim of this report is to illustrate the clinical features of EA-type 5 (EA5) in a chronic migraine Omani patient, with positive family history of migraine and vague attacks of dizziness/vertigo, had no triggering factors and a proved missense mutation in the CACNB4 gene with c.1412G>A (p.Arg471Lys) variant, which has been classified earlier as a variant of uncertain significance.
A 45 years old Omani female, known case of chronic migraine since childhood, presented to our hospital complaining of frequent attacks of vertigo, which started 12 years ago, associated with unsteadiness. At the beginning, she used to have the episodes every two to three months and they were lasting for a maximum of five days. For the last one and a half year, the severity and frequency of her symptoms worsened, getting the episodes almost daily, and became associated with slurred speech and ataxia. The patient had no much improvement to symptomatic treatment.
The patient was evaluated interictally at the neurology outpatient clinic and found to have no history of epilepsy, diplopia, tinnitus, tremor, dystonia or significant weakness and no triggering factor could be identified for her attacks. Her family history was positive for migraine, affecting her grandfather (from paternal side) and all her siblings, as well as attacks of ill-defined vertigo/dizziness, temporarily dissociated with their migrainous headache episodes, not associated with gait disturbance, seizures or abnormal movements, and starting after their third decade of life for all of her female siblings. Based on history, her pedigree chart suggest an autosomal dominant type of inheritance (Figure 1).
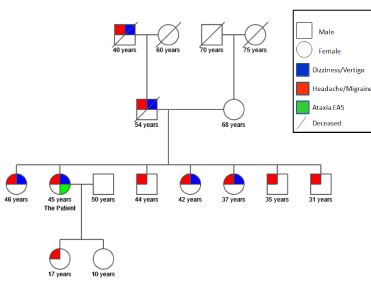
Figure 1. Pedigree of our patient based on history of Headache/Migraine and Dizziness/vertigo
On clinical examination, she had normal cognitive functions, her pupils were reactive to light and accommodation. She has normal extraocular muscles’ movement but with gaze-evoked nystagmus. The patient’s motor examination revealed normal tone and power, in all four limbs, with normal reflexes. Her plantar reflexes were down going and she had no sensory deficit, ataxia, dysarthria, nor other cerebellar signs.
Ictal clinical evaluation showed ataxia with mild dysarthria, prominent horizontal bilateral end-gaze nystagmus and she was unable to perform tandem gait during her attacks. The rest of her cerebellar exam was unremarkable.
Based on the history and clinical grounds, episodic ataxia was highly suspected. The patient’s blood tests were all normal, including complete blood count, renal and liver function tests, and vitamin B12 serum level. Vasculitis and autoimmune screening, as well as her Brain imaging (MRI) results were all normal. Her nerve conduction and electromyography (NCS/EMG) examination were normal and did not show myokymic discharges. The patient was also seen and evaluated by Ear, Nose and Throat (ENT) specialist and found to have normal otology exam.
She was started empirically on Acetazolamide and referred for genetic testing for confirmation of the diagnosis. Whole-exome sequencing test identified a heterozygous variant in the CACNB4 as c.1412G>A (p.Arg471Lys). There was no clinical relevant variant detected in the CACNA1 and therefore, the patient was diagnosed with episodic ataxia type 5 (EA5).
The patient responded very well to treatment with Acetazolamide and her condition greatly improved, in terms of the intensity of her symptoms and the frequency of her episodes. She was also counselled by the genetic centre to screen her siblings as well.
Episodic ataxia is a group of inherited disorders, which manifest as recurrent episodes of incoordination and truncal ataxia, with overlapping clinical features of its’ subtypes [7]. Only EA1 and EA2 have their well-defined clinical features and characteristic interictal findings [6,7,18].
The most common subtype of episodic ataxia is found to be EA 2, which is characterized by recurrent spells of ataxia, interictal nystagmus and slurred speech lasting for hours. Other features, such as seizures, dystonia, tinnitus and cognitive impairment were also reported to occur in EA2 patients [6,19,20]. Both subtypes, EA1 and EA 2, attacks are usually triggered by emotional and physical stressors [1,2,4] and the latter is caused by mutation involving CACNA1A on chromosome 19p13, which encodes Cav2.1, the α1 subunit of the P/Q-type voltage-gated calcium channel [2,6]. Studies have shown that the same genetic mutation is responsible for other conditions, namely familial hemiplegic migraine type 1 (FHM1) and spinocerebellar ataxia type 6 (SCA6); furthermore, there is clinical overlap among these three conditions (EA2, FHM1 and SCA6) [3,7].
On the other hand, EA5 is a rare sub-type that occurs as a result of a mutation in the calcium channel b4 subunit encoding gene CACNB4, on chromosome 2q 22-23 [19,20]. A missense mutation p.Cys 104 phe (C104F) variant has been identified in a French Canadian family with EA5 [8] and other mutations in CACNB4 have been reported in a family with generalized epilepsy (C104F) and in some patients with juvenile myoclonic epilepsy (R482X) [7,8].
EA5 is clinically characterized by recurrent attacks of ataxia accompanied by vertigo, dysarthria, nystagmus, and/or seizures that last for several hours [11,12]. These symptoms are similar to patients with EA2 but tend to begin at a later age, with a typical onset in the third or fourth decades of life [21].
Our case clinches the provisional diagnosis of episodic ataxia in view of her history of episodes of ataxia with normal clinical examination between episodes (apart from gaze-evoked nystagmus), positive family history of similar episodes, the dramatic response to treatment with Acetazolamide and after ruling out other differentials. Labyrinthine abnormalities were ruled out by otolaryngologist examination, both multiple sclerosis and Arnold Chiari malformation were excluded in the view of the clinical examination and MRI results.
The patient is a known case of chronic migraine since childhood, while her progressive spells of imbalance with incoordination started at the age of 33 years. The differential possibility of migraine with brainstem aura (basilar migraine) was ruled out by the absence of other brainstem clinical features, the temporal dissociation between migrainous and ataxic episodes, as well as by the lack of response to all migraine prophylactic therapy. We also excluded episodic ataxia with paroxysmal choreoathetosis and spasticity, as well as familial hemiplegic migraine due to the absence of chorea, stiffness and hemiplegia during headaches’ attacks, although our patient has a family history of migraine.
Migraine association with episodic ataxia is reported in types EA1 and EA2; however, mild generalized headache during the attacks was described in a single patient with EA5 having the well-known mutation (c.311G>T) [22].
EAs are inherited as autosomal dominant disorders and our patient has a significant family history of vertigo/dizziness attacks. Although her family members are not genetically investigated, the pedigree chart showed an autosomal dominant type of inheritance for these attacks. However, not all people with a CACNB4 mutation develop EA5 clinical phenotype and it is estimated that only around 70% of them can have EA5 clinical features [6,23]. This phenomenon is called “reduced or incomplete penetrance” [23].
The reported mutation linked to EA5 is a missense mutation c.311G>T (C104F) variant for CACNB4 on chromosome 2q22-23 [8,19,20]. The same missense mutation (C104F) was identified in a German family with generalized epilepsy [7,8] and another reported CACNB4 mutation was found in some patients with juvenile myoclonic epilepsy (Arg482Ter or R482X) [24]. Our patient’s WES test identified the heterozygote variant in the CACNB4 as c.1412G>A (p.Arg471Lys). This variant causes an amino acid change from Arg. to Lys. at position 471 and it is classified as variant of uncertain significance (class 3) according to the recommendations of Centogene [25] and the American College of Medical Genetics and Genomics (ACMG) guidelines [26].
Many CACNB4 mutation variants have been identified as a “likely benign DNA change” [27,28] but finding this mutation in a symptomatic patient may suggests a likelihood of being associated with EA5 and could explain the absence of coexisting seizures.
Since functional assay and segregation analysis among the patient’s family members could not be done, due to technical reasons, social factors and because of the restrictions of the current health pandemic, we cannot confirm the potential pathogenicity of this variant.
More than 100 different mutations, until now, have been described in all EA genes; however, only 13% to 40% molecular diagnostic rates are reported [29-31]. The majority of genetically confirmed EA have been identified in Caucasian families [29,32-34] and only a few Asians have been reported [7,35,36] ; in addition, a significant proportion of patients with EA do not have mutations in the known EA genes, which supports the genetic heterogeneity of EA and suggests the presence of additional causative genes [7].
Both EA2 and EA5 subtypes respond well to treatment with Acetazolamide which also reduces the frequency of attacks [11,12,19] and our patient responded dramatically well to Acetazolamide, 250 mg 3 times a day. 4-Aminopyridine, a nonselective blocker of voltage-gated potassium channels, is another effective drug in patients with EA2 which can be used for improving the quality of patient’s life by preventing or decreasing the frequency of the attacks [37].
Episodic ataxia is a rare inherited neurological condition; however it’s a treatable entity. Here we report the first case of EA5 in an Omani female patient, known to be a case of chronic migraine, presenting with EA clinical features, no triggering factors and having missense mutation in the CACNB4 gene with c.1412G>A (p.Arg471Lys) variant. We recommend that such patients should undergo a well formed genetic counseling session through which a targeted genetic screening test can be offered to the family members.
Moreover, further studies, including functional assay and segregation analysis among the family members, are needed to prove the potential pathogenicity of this variant and to determine its probable linkage to EA5. Recognition of EA may facilitate the early diagnosis and initiation of treatment. Our patient showed significant improvement with the use of Acetazolamide.
We would like to express our sincere gratitude and appreciation to National Genetic Centre at Royal Hospital, for their valuable contribution.
Informed consent for the publication of the clinical details, test results and radiological images was obtained from the patient and documented in the patient’s electronic file.
The authors declare no financial competing interest. None of the authors received reimbursements, fees, funding, or salary from an organization that may in any way gain or lose financially from the publication of this manuscript. The authors declare no non-financial competing interests (personal, ideological, academic, intellectual, commercial or any other) in relation.
- Baloh RW, Jen JC. Ann NY Acad (2002) Genetics of familial episodic vertigo and ataxia. Sci 956: 338-345.
- Riant F, Vahedi K, Tournier-Lasserve E (2011) Hereditary episodic ataxia. Rev Neurol 167: 401-407.
- Tomlinson SE, Hanna MG, Kullmann DM, Tan SV, Burke D (2009) Clinical neurophysiology of the episodic ataxias: insights into ion channel dysfunction in vivo. Clin Neurophysiol 120: 1768–1776.
- Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, et al. (2007) Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 130: 2484–2493.
- Baloh RW (2012) Handb. Episodic ataxias 1 and 2. Clin Neurol 103: 595-602.
- Kipfer S, Strupp M (2014) The clinical spectrum of autosomal dominant episodic ataxias. Mov Disord Clin Pract 1: 285–290.
- Choi KD, Choi JH (2016) Episodic Ataxias: Clinical and Genetic Features. J Mov Disord 9: 129–135.
- Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, et al. (2000) Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet 66: 1531-1539.
- Jen JC, Wan J, Palos TP, Howard BD, Baloh RW (2005) Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 65: 529-534.
- de Vries B, Mamsa H, Stam AH, Wan J, Bakker SL, et al. (2009) Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol 66: 97-101.
- Damji KF, Allingham RR, Pollock SC, Small K, Lewis KE, et al. (1996) Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias. Arch Neurol 53: 338-344
- Steckley JL, Ebers GC, Cader MZ, McLachlan RS (2001) An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology 57: 1499–1502.
- Kerber KA, Jen JC, Lee H, Nelson SF, Baloh RW (2007) A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol 64: 749-752.
- Conroy J, McGettigan P, Murphy R, Webb D, Murphy SM, et al. (2014) A novel locus for episodic ataxia: UBR4 the likely candidate. Eur J Hum Genet 22: 505-510.
- Gordon N (1998) Episodic ataxia and channelopathies. Brain Dev 20: 9-13.
- Seet RC, Lim EC (2007) A case of intermittent ataxia associated with migraine headaches. CMAJ 177: 565–567.
- Farmer TW, Mustian VM (1963) Vestibulocerebellar ataxia. A newly defined hereditary syndrome with periodic manifestations. Arch Neurol 8: 471-480.
- Hasan SM, D'Adamo MC. Episodic Ataxia Type 1. 2010 Feb 9 [Updated 2018 Nov 1]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Marti S, Baloh RW, Jen JC, Straumann D, Jung HH (2008) Progressive cerebellar ataxia with variable episodic symptoms--phenotypic diversity of R1668W CACNA1A mutation. Eur Neurol 60: 16–20.
- Nachbauer W, Nocker M, Karner E, Stankovic I, Unterberger I, et al. (2014) Episodic ataxia type 2: phenotype characteristics of a novel CACNA1A mutation and review of the literature. J Neurol 261: 983–991.
- Singhvi JP, Prabhakar S, Singh P (2000) Episodic ataxia: a case report and review of literature. Neurol India 48: 78-80.
- González Sánchez M, Izquierdo S, Álvarez S, Bautista Alonso RE, Berciano J, et al. (2019) Clinical manifestations of episodic ataxia type 5. Neurol Clin Pract 9: 503-504.
- Kotagal V (2012) Acetazolamide-responsive ataxia. Semin Neurol 32: 533-537.
- Tadmouri A, Kiyonaka S, Barbado M, Rousset M, Fablet K, et al. (2012) Cacnb4 directly couples electrical activity to gene expression, a process defective in juvenile epilepsy. EMBO J 31: 3730–3744.
- Trujillano D, Oprea GE, Schmitz Y, Bertoli‐Avella AM, Abou Jamra R, et al. A comprehensive global genotype–phenotype database for rare diseases. Mol Genet Genomic Med 5: 66-75.
- Richards S, Aziz N, Bale S, Bick D, Das S, et al. (2015) ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405-424.
- Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, et al. (2011) LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 32: 557-563.
- http://evs.gs.washington.edu/EVS/
- Mantuano E, Romano S, Veneziano L, Gellera C, Castellotti B, et al. (2010) Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J Neurol Sci 291: 30-36.
- Denier C, Ducros A, Vahedi KA, Joutel A, Thierry P, et al. (1999) High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology 52: 1816–1821.
- Eunson LH, Graves TD, Hanna MG (2005) New calcium channel mutations predict aberrant RNA splicing in episodic ataxia. Neurology 65: 308-310.
- Jen JM, Kim GW, Baloh RW (2004) Clinical spectrum of episodic ataxia type 2. Neurology 62: 17-22.
- Maksemous N, Roy B, Smith RA, Griffiths LR. Next-generation sequencing identifies novel CACNA1A gene mutations in episodic ataxia type 2. Mol Genet Genomic Med 4: 211–222.
- Sintas C, Carreño O, Fernàndez-Castillo N, Corominas R, Vila-Pueyo M, et al. (2017) Mutation spectrum in the CACNA1A gene in 49 patients with episodic ataxia. Sci Rep 7: 2514.
- Kim JM, Kim JS, Ki CS, Jeon BS (2006) Episodic ataxia type 2 due to a deletion mutation in the CACNA1A gene in a Korean Family. J Clin Neurol 2: 268–271.
- Hu Y, Jiang H, Wang Q, Xie Z, Pan S (2013) Identification of a novel nonsense mutation p.Tyr1957Ter of CACNA1A in a Chinese family with episodic ataxia 2. PLoS One 8: e56362.
- Strupp M, Kalla R, Dichgans M, Freilinger T, Glasauer S, et al. (2004) Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology 62: 1623-1625.