Using data from the U.S. National Health and Nutrition Examination Surveys, 1999–2014, we tested the hypothesis that excess insulin and hypoxia might be catalysts for obesity. An empirical model of obesity was constructed using excess insulin and hypoxia as mediators between excess nutrients and obesity. Excess nutrients upregulate excess insulin, which in turn upregulates intracellular storage of nutrients. Depending on oxygen supply and energy demands, intracellular excess nutrients are processed via [1] aerobic cellular respiration (producing energy), [2] lipogenesis (leading to obesity), [3] glycation (producing advanced glycation end products), or [4] lactic acid fermentation (producing lactic acids). One quantile jump in insulin increased the likelihood for obesity; hypoxia did so as well, but with a lesser magnitude. The intercorrelation between insulin and hypoxia was significant. Prescription medications and excess nutrients had significant positive effects on insulin and hypoxia. Exercise showed a positive relationship with excess insulin but a negative one with hypoxia. The findings confirmed our hypothesis that excess insulin and hypoxia were mediators for obesity. Strategies to control obesity should focus on preventing excess insulin and hypoxia through careful lifestyle modifications such as diet and aerobic exercises before medical or pharmacological therapy.
excess insulin, hypoxia, obesity, type 2 diabetes, glycation
Obesity, a pandemic in the U.S. [1], is continuing to worsen. In 2013–2014, more than a third of American adults (37.7%) were obese, with a body mass index (BMI) of ≥30. That figure represents an increase of 7.2% from the years 1999–2000, roughly equivalent to 16 million people. During the same period in the U.S., the prevalence of the morbidly obese (BMI ≥40) increased from 4.7% to 7.7% in 2013–2014, representing 6.7 million. Perhaps not surprisingly, bariatric surgery grew from 158,000 cases in 2011 to 216,000 in 2016 [2].
Obesity, the accumulation of excess body fat, is linked to many chronic conditions or diseases as a cause of comorbidities, which include asthma [3], type 2 diabetes [4,5], rheumatoid arthritis [6], cardiovascular diseases [7,8], cancer [9], depression [10,11], and dementia [12,13]. On average, an obese person incurred $3,429 more healthcare costs (in 2013 dollars) from 2006 to 2013 than did a non-obese person [14]. Beyond that, the social costs of obesity are incalculable.
Despite enormous efforts to curtail the rising trend in obesity, the war against obesity is losing its footing, with no clear strategy to overcome this challenge in sight [15]. This is not because the causes of obesity are unknown; the WHO’s International Statistical Classification of Diseases and Related Health Problems version 10 (ICD-10) identified excess calories and drugs as obesity’s most important causes. But homocentric theories of obesity at both macro and micro levels seem to have prevailed and misguided the practice of obesity control. At the macro level, for example, the homeostatic theory of obesity has suggested “a circle of discontent” linking weight gain, body dissatisfaction, negative affect, and overconsumption, resulting in strategies to overcome the obesity epidemic such as putting a stop to victim-blaming, stigma, and discrimination and revalorizing the thin ideal, among others [16]. At the micro level, extensive studies of genetic and biochemical mechanisms have linked excess nutrients to obesity as a byproduct of metabolism, but the linking mechanisms are still poorly understood. This may be because the genetic and molecular processes of metabolism are complex and complicated, but it may also be that a homocentric misinterpretation of the role of insulin, one of the most critical hormones in metabolism, has prevailed in research.
It is well known that insulin regulates uptake of nutrients (glucose, fatty acids, and amino acids) [17]. Further, insulin concentration modulates hepatic lipogenesis in mice [18], indicating that excess insulin could lead to obesity. Nevertheless, insulin has been regarded as nonessential to obesity [19], with obesity-induced insulin resistance leading to type 2 diabetes [20,21], or simply as part of a continual, concurrent progression along with obesity. In the carbohydrate–insulin model of obesity [22] that identified insulin as a cause of obesity, insulin resistance works selectively to make non-adipocytes starve and adipocytes fat at the same time [23]. In this paper, we present a series of analyses of a national dataset to demonstrate the mediating role of insulin and oxygen for obesity. We also discuss the implications and applicability of this knowledge for preventing and managing obesity and obesity-related chronic conditions.
Theoretical framework
We hypothesize that the linking mechanisms between the causes identified by the ICD-10 (e.g., excess calories and drugs) and obesity are two mediators: (a) excess insulin and (b) hypoxia. These mediators are catalysts for the transformation of nutrients to energy through cellular respiration. Figure 1 schematically depicts this relationship.
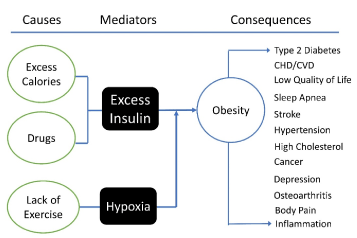
Figure 1. Theoretical framework of obesity, in which excess insulin and hypoxia mediate personal factors (excess calories, drugs and sedentary lifestyle) for obesity, which in turn leads to other physical and/or mental problems. Because obesity is a continuous process, there can be a feedback loop that may reinforce/accelerate the process, but the effects of the feedback loop can be negligible.
Insulin as keeper
Vertebrate insulin is a peptide hormone secreted by pancreatic beta cells. Insulin has 51 amino acid residues in two chains; the molecular structures of vertebrate insulins are so similar that they are clinically exchangeable [24]. Insulin’s simple structure and universal function indicate that it evolved to perform a specific and essential function - to store nutrients [25]. In addition, insulin is structurally and functionally robust against mutations; only three cases of nonsense mutant human insulin have been reported since the 1960s [26,27].
Because insulin is such a potent hormone, it is tightly controlled and regulated. Insulin secretion is influenced by plasma glucose, which is in turn affected by food intake, autonomic circadian rhythm, neurological responses such as memory, or physiological conditions such as acute and chronic illnesses or exercise [28-30]. When glucose enters the portal vein, the pancreatic beta cells secrete and release insulin into the bloodstream almost immediately. Pancreatic glucose-sensing mechanisms in carnivores and non-carnivores are fundamentally similar [31]. The pancreatic glucose sensor detects the glucose level, which triggers a cascade of insulin secretion into the portal vein accordingly [32]. Further, because fat-sensing receptors exist in pancreatic beta cells [33], secretion increases at the presence of fatty acids and possibly other nutrients (e.g., amino acids, triglycerides). It is therefore reasonable to assume that the amount of plasma insulin is proportional to that of plasma nutrients, which include not only glucose but also fatty acids, and possibly amino acids.
Plasma nutrients must be transferred from the bloodstream and the interstitial space to intracellular space as quickly as possible; prompt processing may be mandatory to reduce the chances for glycation [34,35]. Through the action of insulin, they are stored in three different types of cells for different time periods: (a) adipocytes, for days or months; (b) hepatocytes, for several hours; (c) all other cells, for a few minutes. The exact mechanism of allocating nutrients into the three storage compartments is unknown, but factors including the anabolic capacity of hepatocytes (e.g., glycogenesis and lipogenesis), excess nutrients, and the cell’s energy demands may be involved. Because insulin increases the intracellular uptake of glucose and fatty acids by increasing the number of glucose transporters (e.g., GLUT4) [36] and fatty acid transporters [37], all nutrients (glucose, fatty acids, and possibly amino acids) are stored regardless of cell types.
If excess nutrients enter the portal vein, surplus nutrients pass through the liver to be released directly into the bloodstream while further increasing insulin secretion. Through the increased action of insulin, more nutrients enter the intracellular compartments, possibly along with the insulin and the insulin receptor complex [38].
Hypoxia in cell respiration
Once nutrients move into cells other than adipocytes or hepatocytes, they cannot return to the intercellular space or bloodstream. Oxygen is so critical in cell respiration that it must be continually supplied. If oxygen supply stops for several minutes, cell destruction (i.e., death) begins. Living organisms do not carry extra oxygen tanks, because oxygen is abundant and ubiquitous. But the omnipresent abundance of oxygen does not imply that it is less significant than excess nutrients are for obesity, for which foods are scarce and sporadic. The impacts of hypoxia or hyperoxia can be deadly and immediate.
Several factors influence the oxygen supply to cells, including O2 and CO2 concentration, plasma acidity, and temperature [39]. Besides respiratory system dysfunctions (e.g., alveolar hypoventilation), hemoglobin health is critical because hemoglobin carries oxygen to tissues. If plasma glucose is attached to hemoglobin (i.e., glycated), the affected hemoglobin is no longer able to move oxygen because of glucose’s affinity to oxygen: It holds the oxygen to the iron (Fe) receptacle forever, and the captured oxygen is not released, regardless of CO2 concentration. Therefore, oxygen supply is inversely proportional to glycated hemoglobin (i.e., hemoglobin A1c, or A1C for short), because the glycation rate is constant over time [40]. The higher the A1C, the less the oxygen delivered to cells, resulting in de facto hypoxia.
A glucose molecule enters glycolysis, which generates 2 adenosine triphosphate (ATP) molecules and 2 pyruvates, and this process does not require oxygen in the cytosol. Pyruvate enters the mitochondria into the citric acid cycle (i.e., cell respiration) when oxygen is sufficient. Fatty acids go through the more complicated process to become acetyl coenzyme A (acetyl-CoA) via beta oxidation to enter the citric acid cycle [41], which requires more oxygen.
Under the hypoxic condition when energy demand is low, excess acetyl-CoA is put back into lipogenesis to synthesize triglycerides [42], which is most likely to increase the cell volume and weight. When energy demand increases, the pyruvate goes through lactic acid fermentation. This process not only generates less energy but also produces lactic acids, a deadly combination that leads to several problems: “(a) collapse of Na+ and K+ gradients, (b) depolarization of membranes, (c) cellular uptake of Cl−, (d) cell swelling, (e) increased cytosolic Ca2+ concentration, and finally, (f) decreased cytosolic pH, resulting in intracellular acidosis in tumor cells” [43, p. 267].
Sample selection and analysis
This report is a secondary analysis of data from the annual National Health and Nutrition Examination Survey (NHANES), the most comprehensive assessment of the nation’s health status [44]. To increase the power and precision of estimates, we combined the 8 cycles of surveys from 1999 through 2014, using the appropriate sampling weights: For data extraction and management, including missing observations, we followed the official guideline [45]. We used STATA (version 14; College Station, TX) for analysis, with P < 0.05 considered significant in two-tailed tests.
Measurements
The NHANES provides BMI (kg/m2) as a proxy variable for obesity. We excluded children (age <20) from our analysis, because their BMI can be interpreted differently than adults’ [45]. The NHANES reported fasting plasma insulin (hereafter “insulin”), endogenous and exogenous combined, in uU/ml and pmol/L from a total of 24,963 persons. For analysis, we used insulin measured in pmol/L and distributed in quantiles.
A1C was used as an indicator of hypoxia. For example, 6.0% A1C can deliver 3% more oxygen to target cells than can 9.0% A1C. This 3% makes a huge difference, because a person breathes on average more than 30,000 times a day. Higher A1C results in a shortage of oxygen—hypoxia. The NHANES reports A1C as a percentage of glycated hemoglobin in red cells. We used the actual values in percentages as well as distributed in quantiles in our analysis.
Energy from fat, as a proxy of excess calories, ranged from 0% to 93%. We used its distribution in quantiles in our analysis. Daily exercise was calculated from a series of questions and reclassified into four categories (0 = no daily exercise, 1 = less than 30 minutes, 2 = less than 60 minutes, 3 = more than or equal to 60 minutes). The number of prescription drugs, extracted from interview questions on drugs, ranged from no prescription drugs to 23 different drugs, with a mean of 1.9 drugs in the whole population of age 21 or older, and 3.3 drugs among those who were taking prescription drugs. Those who were taking more than 3 medications were combined into one group, which resulted in four categories (0 = none, 1 = one drug, 2 = two drugs, 3 = three drugs or more).
Obesity distribution
From 1999 through 2014, about one third (32.2%) of Americans age 21 or older were normal in body weight, another third (34.1%) were overweight, and the remaining third were obese (28.0%) or morbidly obese (5.9%). The BMI distribution by sex was bell-shaped, with a peak for age at 50s in women and 60s in men and a valley at 80s for both genders. In each age group (in 10s), BMI was not statistically different between women and men. A similar bell-shaped BMI distribution was found for race and age, but it was highest in Blacks, followed by Mexican Americans, Whites, and others. The differences were statistically significant for each age group except those in their 70s and 80s (Figure 2).
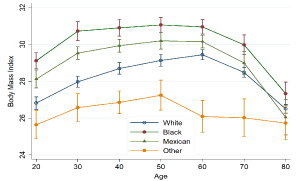
Figure 2. Body Mass Index distribution by race and age (in 10s), with 95% confidence intervals. (Data source: National Health and Nutrition Examination Survey, 1999–2014.)
Factors associated with obesity
Obesity, measured by ordered categories of BMI (normal, <25; overweight, ≥ 25; obese, ≥ 30; morbidly obese, ≥ 40), was significantly correlated with several variables among demographic characteristics, metabolism-related biomarkers, diet and exercise, and hematological characteristics. Those with normal BMI were younger than the overweight or obese. Males were more overweight or obese than females, but less morbidly obese. Compared with Whites, Blacks and Mexican Americans were more overweight or obese; Blacks were more morbidly obese than Whites. Others, including Asian Americans, were less overweight or obese than Whites. Education level and family income were inversely correlated with obesity.
Metabolism-related biomarkers were positively correlated with obesity; these included systolic and diastolic blood pressure, blood sugar, A1C, total cholesterol, C-peptide (the prohormone to insulin), insulin, and C-reactive protein as a marker of inflammation. Among these, insulin and A1C were prominent (Figure 3). Both insulin and A1C were proportional to obesity, but insulin was stable across all age groups, whereas A1C tended to increase with age.
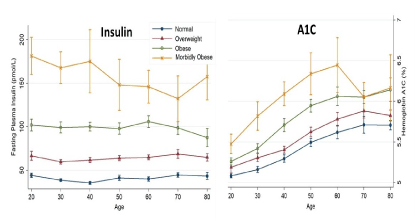
Figure 3. Distribution of insulin (left) and hemoglobin A1C (right) by obesity status and by age (in 10s) with 95% confidence intervals. (Data source: National Health and Nutrition Examination Survey, 1999–2014.)
Schematic path model of obesity
Based on our proposed theoretical framework, several variables were included in a path model to explain obesity; their relationships are schematically depicted in Figure 3. We ran a series of ordered logistic regressions and obtained odds ratios with standard errors. All relationships were statistically significant, and the magnitudes and directions were as expected. For example, persons in the higher quantile for insulin were more likely to be obese than persons in the lower quantile. The likelihood of being overweight or obese increased exponentially when quantiles were compared with the 1st quantile for insulin: For persons in the 2nd quantile, OR = 2.90, 95% CI: 2.52, 3.24, t = 18.8, P < 0.001; for persons in the 3rd quantile, OR = 7.52, 95% CI: 6.78, 8.34, t = 38.7, P < 0.001; and for persons in the 4th quantile, OR = 24.86, 95% CI: 21.61, 28.61, t = 45.4, P < 0.001.
Similar significant relationships were observed between hypoxia and obesity, but the magnitudes were smaller than those for insulin: OR = 1.32, 95% CI: 1.27, 1.37, t = 14.4, P < 0.001. Compared with persons in the 1st quantile of hypoxia, persons in higher quantiles had greater chances of being obese: For persons in the 2nd quantile, OR = 1.37, 95% CI: 1.25, 1.50, t = 6.8, P < 0.001; for persons in the 3rd quantile, OR = 1.65, 95% CI: 1.48, 1.84, t = 9.1, P < 0.001; and for persons in the 4th quantile, OR = 2.13, 95% CI: 1.89, 2.40, t = 12.4, P < 0.001. Further, fat consumption as a proxy of excess calories and the number of prescription drugs were positively correlated with insulin and hypoxia. The magnitude of these relationships was modest in comparison with those of insulin and hypoxia to obesity, except for the relationship of prescription drugs to hypoxia, OR = 1.42, t = 32.0, 95% CI: 1.39, 1.45, P < 0.001. Exercise decreased the insulin level but increased hypoxia as expected.
The results of this analysis show that excess insulin coupled with hypoxia is a significant catalyst for obesity. In addition, excess nutrients and prescription drugs significantly increased both insulin and hypoxia levels. By focusing on the primary function of insulin, our model’s elucidation of the process of obesity is consistent with empirical data: Excess nutrients relative to the energy demands and the oxygen supply in all cells are likely to increase the chances of (1) glycogenesis and lipogenesis, increasing cell weight and volume; (2) glycation, resulting in increase in advanced glycation end products (AGEs); and (3) lactic acid fermentation, resulting in reduced ATP but increases in lactic acids. This process occurs in cytosol of all cells that generate energy for their survival and operation.
The propensity of each mechanism under the condition of intracellular excess nutrients and oxygen shortage is yet to be elucidated; we suspect that the number of mitochondria and the intercellular energy demand may be factors. For example, in a hepatic cell that has mitochondria in thousands but requires a low level of energy in comparison with a muscle cell, glycogenesis and lipogenesis are more likely to occur. In a red blood cell that requires high energy for movement but relies on glycolysis and fermentation due to the lack of mitochondria, glycation is likely to prevail, as is evidenced by the high level of glycation in red blood cells (i.e., hemoglobin A1c). In white adipocytes, which require much less energy and where the number of mitochondria is small, lipogenesis can prevail. In muscular skeletal cells, which require a high level of energy, all three mechanisms may be possible.
The first mechanism directly contributes to obesity by accumulating triglycerides, which can have consequential effects by increasing the volume and weight of adipocytes but minimal effects on other cells because they are more stable than lipids and glycerol in their bioactivities and they can be transferred to adipocytes. On the contrary, the second mechanism (glycation) coupled with the third (fermentation) can compromise cellular function by accumulating AGEs [34]. Glycation itself is slow, owing to the infinitesimal size of glucose and protein molecules in cytosol, but over time it can accumulate enough AGEs, leading to the obesity-related complications listed in Figure 1. Our model consistently explains the obesity process and obesity-related complications by unifying the most important two catalysts of metabolism. For example, the model is consistent with the process of type 2 diabetes that consists of glycation and fermentation due to excess insulin and a shortage of oxygen. The process leads to clinical indications including elevated A1C, swelling, inflammation, low body temperature in extremities, infection, peripheral nerve damage, loss of short-term memory, and depressive mood (Figure 4).
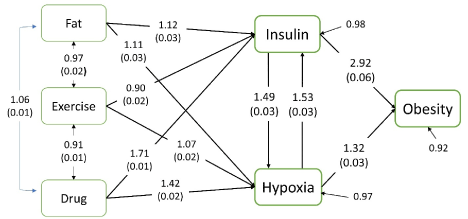
Figure 4. Schematic path diagram leading to obesity. Coefficients are odds ratios of quantiles with linearized standard errors in parenthesis. All coefficients are statistically significant at P < 0.001, except between Fat and Exercise. (Data source: National Health and Nutrition Examination Survey, 1999–2014.)
The causes of both obesity and type 2 diabetes are the same: excess insulin and hypoxia. Obesity, however, can manifest itself within several months, whereas type 2 diabetes develops over decades, because lipogenesis is much faster than glycation. Therefore, the key to controlling both obesity and type 2 diabetes is to control excess insulin and hypoxia, which slows lipogenesis and glycation and can eventually reverse the process of obesity and type 2 diabetes.
The NHANES data in Figure 5 illustrate the need for controlling excess insulin to manage type 2 diabetes. People diagnosed with type 2 diabetes are classified into several groups according to combinations of three types of diabetes drugs in addition to a no-medication group: (a) metformin, (b) oral medications other than metformin, and (c) insulin. The levels of glucose and insulin in the normal (without diabetes/prediabetes and not on antidiabetic medication) are used as a reference point at 100%, where the difference between glucose and insulin levels is 0%. The levels of glucose and insulin in the no-medication group are 147% and 138%, respectively, a difference of +9% (i.e., more glucose); for metformin only, 150% and 144%, respectively, with a difference of +6%; and for oral drugs only, 146% and 144%, with a dfference of +2%. The relationships between glucose and insulin in these groups are consistent and stable, although both glucose and insulin are much higher than normal. However, when insulin therapy is involved, the relationship is inconsistent and unstable. In the insulin-only group, the glucose and insulin levels jump to 188% and 303%, respectively, with a difference of -115%; in the group with metformin plus insulin, the levels are 191% and 334%, with a difference of -143%. This figure clearly demonstrates that the detrimental effects of antidiabetic drugs are mediated by excess insulin, which explains the 2–3 times higher risks of cardiovascular mortality among those who receive antidiabetic medicines [46].
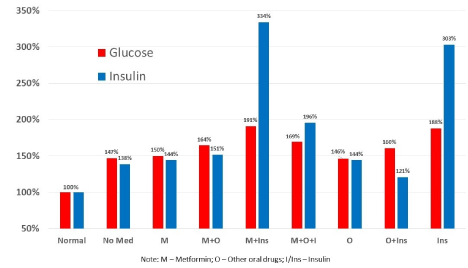
Figure 5. Fasting plasma glucose and insulin level by diabetes medication status in the U.S. population age 21 or older; normal glucose and insulin at 100%. (Data source: National Health and Nutrition Examination Survey, 1999–2014.)
Our findings explicate the linking mechanisms between excess nutrients and obesity and type 2 diabetes. We have intentionally avoided using the term insulin resistance, which is a homocentric misnomer for excess insulin. Since the first observation that insulin resistance manifested itself only among people who had used exogenous insulin (i.e., insulin injections) for months [47], insulin resistance remained hypothetical and yet to be proven [48]. Further, insulin resistance cannot be measured with any direct measure (e.g., insulin itself, insulin antibodies, or something else detrimental to insulin’s action) but only indicated by an indirect measure (i.e., elevated glucose level), which is truly ironic in an age of advanced measurement technologies. Still worse, even though measurement of plasma insulin has been readily available for decades, confusion regarding insulin release by the pancreas beta cells remain. For example, in a review in Nature, Kahn, Hull, and Utzschneider [49] compared the amount of insulin release and insulin sensitivity among several groups in comparison with healthy adults. The first figure in that article indicates that the insulin release for those with type 2 diabetes was about 20% of that for healthy adults [p. 841]. The authors provided no data source for the data in the figure, and indeed the figure was incorrect. Ever since plasma insulin was first measured by Berson and Yalow [50], type 2 diabetes, formerly known as non-insulin-dependent diabetes mellitus, has been indicated by the elevated levels of both insulin and glucose (Figure 5).
In sum, our findings suggest effective strategies to control obesity and type 2 diabetes that lower plasma insulin by reducing nutrient intake and that improve oxygen supply by aerobic exercise. We have incorporated this strategy into our community-based type 2 diabetes management programs and demonstrated its effectiveness with significant, sustained reductions of A1C, blood pressure, and depression while improving quality of life [51].
This work was partially supported by the Technology Innovation Program (or Industrial Strategic Technology Development Program (10053704, Development of Smart Healthcare System and Pilot Project for Military Personnel and Global Healthcare) funded By the Ministry of Trade, Industry & Energy (MOTIE, Korea). Editorial support with manuscript development was provided by the Cain Center for Nursing Research and the Center for Transdisciplinary Collaborative Research in Self-Management Science (P30, NR015335) at The University of Texas at Austin School of Nursing.
Drs. K. B. and M. T. Kim had full access to the data and wrote the manuscript.
- Ogden CL, Carroll MD, Fryar CD, Flegal KM (2015) Prevalence of Obesity Among Adults and Youth: United States, 2011-2014. NCHS Data Brief : 1-8. [Crossref]
2021 Copyright OAT. All rights reserv
- Estimate of bariatric surgery numbers, 2011-2016 (2016) American Society of Metabolic and Bariatric Surgery.
- Kanagalingam S, Dixon AE (2015) The asthma–obesity epidemic. Prim Prev Insights 5: 11-17.
- McCarthy MI (2010) Genomics, type 2 diabetes, and obesity. N Engl J Med 363: 2339-2350. [Crossref]
- Cohen DH, LeRoith D (2012) Obesity, type 2 diabetes, and cancer: the insulin and IGF connection. Endocr Relat Cancer 19: F27-45. [Crossref]
- Liu Y, Hazlewood GS, Kaplan GG, Eksteen B, Barnabe C (2017) Impact of obesity on remission and disease activity in rheumatoid arthritis: a systematic review and meta-analysis. Arthritis Care Res 69:157-165. [Crossref]
- De Schutter A, Lavie CJ, Milani RV (2014) The impact of obesity on risk factors and prevalence and prognosis of coronary heart disease—the obesity paradox. Prog Cardiovasc Dis 56: 401-408. [Crossref]
- Lavie CJ, McAuley PA, Church TS, Milani RV, Blair SN (2014) Obesity and cardiovascular diseases: implications regarding fitness, fatness, and severity in the obesity paradox. J Am Coll Cardiol 63: 1345-1354. [Crossref]
- Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, et al. (2016) Body Fatness and Cancer--Viewpoint of the IARC Working Group. N Engl J Med 375: 794-798. [Crossref]
- Preiss K, Brennan L, Clarke D (2013) A systematic review of variables associated with the relationship between obesity and depression. Obes Rev 14: 906-918. [Crossref]
- Papazoglou IK, Jean A, Gertler A, Taouis M, Vacher CM (2015) Hippocampal GSK3β as a Molecular Link Between Obesity and Depression. Mol Neurobiol 52: 363-374. [Crossref]
- Hildreth KL, Van Pelt RE, Schwartz RS (2012) Obesity, insulin resistance, and Alzheimer's disease. Obesity (Silver Spring) 20: 1549-1557. [Crossref]
- Kiliaan AJ, Arnoldussen IA, Gustafson DR (2014) Adipokines: a link between obesity and dementia? Lancet Neurol 13: 913-923. [Crossref]
- Biener A, Cawley J, Meyerhoefer C (2017) The High and Rising Costs of Obesity to the US Health Care System. J Gen Intern Med 32: 6-8. [Crossref]
- Ng M, Fleming T, Robinson M, et al. (2014) Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384: 766-781. [Crossref]
- Marks DF (2015) Homeostatic theory of obesity. Health Psychol Open 2: 2055102915590692. [Crossref]
- White MF, Kahn CR (1993) Mechanisms of insulin action. In: Moller DE, ed. Insulin Resistance. Chichester, UK: Wiley; pp: 9-47.
- Softic S, Kirby M, Berger NG, Shroyer NF, Woods SC, et al. (2012) Insulin concentration modulates hepatic lipid accumulation in mice in part via transcriptional regulation of fatty acid transport proteins. PloS One 7: e38952. [Crossref]
- Basman CL (2018) Obesity. In: Ferrie FF, ed. 2018 Ferrie’s Clinical Advisor. Philadelphia, PA: Elsevier; pp: 906-909.
- Shelton RC, Miller AH (2010) Eating ourselves to death (and despair): the contribution of adiposity and inflammation to depression. Prog Neurobiol 91: 275-299. [Crossref]
- Pendergrass M, Bertoldo A, Bonadonna R, et al. (2007) Muscle glucose transport and phosphorylation in type 2 diabetic, obese nondiabetic, and genetically predisposed individuals. Am J Physiol Endocrinol Metab 292: E92-E100. [Crossref]
- Luchsinger JA (2008) Adiposity, hyperinsulinemia, diabetes and Alzheimer's disease: an epidemiological perspective. Eur J Pharmacol 585: 119-129. [Crossref]
- Hall KD (2017) A review of the carbohydrate-insulin model of obesity. Eur J Clin Nutr 71: 323-326. [Crossref]
- Mayer JP, Zhang F, DiMarchi RD (2007) Insulin structure and function. Peptide Sci 88: 687-713. [Crossref]
- Herring R, Jones RH, Russell-Jones DL (2014) Hepatoselectivity and the evolution of insulin. Diabetes Obes Metab 16: 1-8. [Crossref]
- Tager H, Given B, Baldwin D, Mako M, Markese J, et al. (1979) A structurally abnormal insulin causing human diabetes. Nature 281: 122-125.
- Sakura H, Iwamoto Y, Sakamoto Y, Kuzuya T, Hirata H (1986) Structurally abnormal insulin in a diabetic patient: characterization of the mutant insulin A3 (Val----Leu) isolated from the pancreas. J Clin Invest 78: 1666-1672. [Crossref]
- MacDonald PE, Joseph JW, Rorsman P (2005) Glucose-sensing mechanisms in pancreatic beta-cells. Philos Trans R Soc Lond B Biol Sci 360: 2211-2225. [Crossref]
- Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, et al. (2015) Pancreatic β cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science 350: aac4520. [Crossref]
- Sadacca LA, Lamia KA, deLemos AS, Blum B, Weitz CJ (2011) An intrinsic circadian clock of the pancreas is required for normal insulin release and glucose homeostasis in mice. Diabetologia 54: 120-124. [Crossref]
- Schermerhorn T (2013) Normal glucose metabolism in carnivores overlaps with diabetes pathology in non-carnivores. Front Endocrinol (Lausanne) 4: 188. [Crossref]
- Han K, Kang H, Choi MY, Kim J, Lee MS (2012) Mathematical model of the glucose–insulin regulatory system: from the bursting electrical activity in pancreatic β-cells to the glucose dynamics in the whole body. Phys Lett A 376: 3150-3157.
- Priyadarshini M, Wicksteed B, Schiltz GE, Gilchrist A, Layden BT (2016) SCFA receptors in pancreatic β cells: novel diabetes targets? Trends Endocrinol Metab 27: 653-664. [Crossref]
- Salahuddin P, Rabbani G, Khan RH (2014) The role of advanced glycation end products in various types of neurodegenerative disease: a therapeutic approach. Cell Mol Biol Lett 19: 407-437. [Crossref]
- Szwergold BS (2005) Intrinsic toxicity of glucose, due to non-enzymatic glycation, is controlled in-vivo by deglycation systems including: FN3K-mediated deglycation of fructosamines and transglycation of aldosamines. Med Hypotheses 65: 337-348. [Crossref]
- Bogan JS (2012) Regulation of glucose transporter translocation in health and diabetes. Annu Rev Biochem 81: 507-532. [Crossref]
- Buqué X, Cano A, Miquilena-Colina ME, García-Monzón C, Ochoa B, et al. (2012) High insulin levels are required for FAT/CD36 plasma membrane translocation and enhanced fatty acid uptake in obese Zucker rat hepatocytes. Am J Physiol Endocrinol Metab 303: E504-E514. [Crossref]
- Duckworth WC, Bennett RG, Hamel FG (1998) Insulin degradation: progress and potential. Endocr Rev 19: 608-624. [Crossref]
- Pittman RN (2016) Regulation of Tissue Oxygenation. (2nd Edn) San Rafael, CA: Morgan & Claypool.
- Ladyżyński P, Wójcicki JM, Bąk MI, Sabalińska S, Kawiak J, et al. (2011) Hemoglobin glycation rate constant in non-diabetic Individuals. Ann Biomed Eng 39: 2721-2734. [Crossref]
- Houten SM, Wanders RJ (2010) A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis 33: 469-477. [Crossref]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, et al. (2011) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481: 380-384. [Crossref]
- Höckel M, Vaupel P (2001) Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Nat Cancer Inst 93: 266-276. [Crossref]
- Johnson CL, Paulose-Ram R, Ogden CL, et al. (2013) National Health and Nutrition Examination Survey: Analytic Guidelines, 1999–2010. Vital Health Stat 1-24. [Crossref]
- Llewellyn A, Simmonds M, Owen CG, Woolacott N (2016) Childhood obesity as a predictor of morbidity in adulthood: a systematic review and meta-analysis. Obes Rev 17: 56-67. [Crossref]
- Gamble JM, Simpson SH, Eurich DT, Majumdar SR, Johnson JA (2010) Insulin use and increased risk of mortality in type 2 diabetes: a cohort study. Diabetes Obes Metab 12: 47-53. [Crossref]
- Yalow RS, Berson SA (1961) Immunologic aspects of insulin. Am J Med 31: 882-891. [Crossref]
- Erion DM, Shulman GI (2010) Diacylglycerol-mediated insulin resistance. Nature Med 16: 400-402. [Crossref]
- Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444: 840-846. [Crossref]
- Berson SA, Yalow RS (1966) Insulin in blood and insulin antibodies. Am J Med 40: 676-690. [Crossref]
- Kim MT, Kim KB, Huh B, Nguyen T, Han HR, et al. (2015) The Effect of a Community-Based Self-Help Intervention: Korean Americans With Type 2 Diabetes. Am J Prev Med 49: 726-737. [Crossref]