Rhabdomyomas are the most common cardiac tumours and are easily diagnosed by echocardiography. Heart failure and arrhythmias are two major complications of rhabdomyomas in foetal life and in newborns. We report the case of an apparently healthy male newborn with sporadic ventricular premature beats. The echocardiographic evaluation performed at birth showed multiple rhabdomyomas with a significant obstruction of the left and right ventricular outflow tracts. During the follow-up, we observed absence of neurological problems, no signs of heart failure, and normalization of the arrhythmic burden. Serial echocardiographic evaluation showed a reduction of the size and number of rhabdomyomas. The subsequent 14-year follow-up was completely uneventful. In conclusion, owing to the favourable natural history of rhabdomyomas, most patients can be managed conservatively.
Cardiac tumours are a rare congenital condition and can be easily detected by echocardiography during foetal life [1] or at birth [2]. The most common lesion during infancy and childhood, accounting for 50-75% of paediatric cardiac tumours, is rhabdomyoma, a hamartomatous lesion arising from cardiac myocytes [3]. Approximately 51-86% of children diagnosed with cardiac rhabdomyomas have a positive family history or demonstrate clinical or radiologic evidence of tuberous sclerosis [4].
Heart failure and arrhythmias are two major complications of rhabdomyomas in foetal life and in newborns. Large masses interfering with cardiac valves or obstructing the left (LVOT) or right ventricular outflow tract (RVOT) may provoke heart failure and death. Life-threatening arrhythmias may be triggered by two different mechanisms. First, rhabdomyoma fibres may be used as accessory pathways for incessant supraventricular re-entry tachycardia; second, they may represent foci for automatic sustained ventricular tachycardia (VT) [5,6]. Nevertheless, the natural history of cardiac rhabdomyomas is usually favourable, with complete or partial regression and subsequent resolution of symptoms. The reported survival rates range from 81% to 92% [5,6].
In April 2006, an apparently healthy male newborn was diagnosed to have signs of biventricular hypertrophy and sporadic ventricular premature beats of two morphologies, originating from the left ventricular apex and the RVOT. No foetal echocardiographic evaluation was accomplished at that time. The echocardiographic evaluation performed shortly after birth showed numerous large “echodense” masses localized intramurally in the left and right ventricle, compatible with multiple rhabdomyomas. The cavity was completely obliterated by three large masses localized at the left ventricular apex (Figure 1). Left and right ventricular mid cavities were almost entirely occupied by other masses (Figure 2). A large mass caused a significant obstruction of the LVOT with an intraventricular gradient of 47 mmHg (Figure 3). Another lesion, detected in the RVOT very close to the pulmonary valve, was responsible for a mild flow acceleration without significant gradient (Figure 4). The mitral pattern at pulsed wave Doppler evaluation showed signs of impaired left ventricular diastolic function (Figure 5).
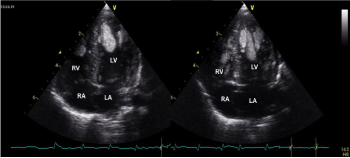
Figure 1. Three large masses localized at the left ventricular apex, with complete cavity obliteration
LA: left atrium; LV: left ventricle; RA: right atrium, RV: right ventricle
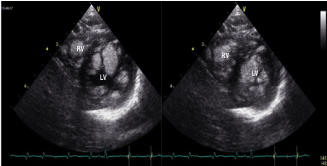
Figure 2. Left and right ventricular mid cavities almost completely occupied by multiple rhabdomyomas
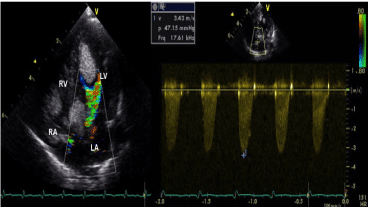
Figure 3. A large mass causing a significant obstruction of the left ventricular outflow tract (A) with an intra-ventricular gradient of 47 mmHg (B)
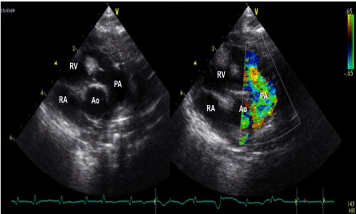
Figure 4. A large rhabdomyoma detected in the right ventricular outflow tract, very close to the pulmonary valve, responsible for mild flow acceleration without significant gradient
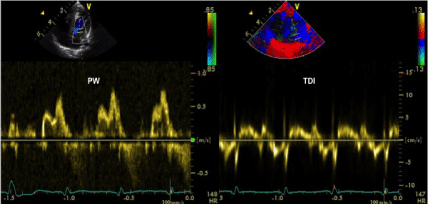
Figure 5. Anomalous mitral pattern at pulsed wave Doppler evaluation showing signs of impaired left ventricular diastolic function
24-h Holter ECG monitoring showed about 1500 isolated premature ventricular beats of two morphologies.
There was no family history of tuberous sclerosis or rhabdomyomatosis. At 2 months of life, the patients underwent cerebral and cardiac magnetic resonance imaging, which was negative for brain masses due to tuberous sclerosis and confirmed the presence of numerous intramural masses in the left and right ventricle, compatible with multiple rhabdomyomas. Genetic testing excluded pathogenic variants of the TSC1 and TSC2 genes, that are associated with tuberous sclerosis complex.
After few weeks, the patient was discharged and followed very strictly in our outpatient programme. During the follow-up, we observed a normal growth, absence of neurological problems, no signs of heart failure, and normalization of the arrhythmic burden at different 24-h Holter ECG monitoring. Starting from the sixth month of life, serial echocardiographic evaluation showed a progressive reduction of the size and number of multiple rhabdomyomas in both ventricles simultaneously, with a decrease of the LVOT gradient. Considering the favourable course of the disease and the risks related to the resection of large cardiac tumours, in agreement with cardiac surgeons we opted for a watchful waiting approach. At 3-year follow-up, complete remission of the masses was achieved, and echocardiographic findings were essentially normal. The subsequent 14-year follow-up was completely uneventful.
The growth of rhabdomyomas in foetal life occurs mainly between the second and third trimester, slows towards the end of pregnancy [7] and often ends after birth, probably due to the absence of pregnancy hormones [8]. Fesslova et al. [1] observed that some small masses grow proportionally, remaining quite stable throughout gestation, while larger masses may have a disproportional growth causing a partial haemodynamic obstruction of the RVOT and LVOT. Rhabdomyomas are easily diagnosed by echocardiography during foetal life and at birth. They appear on as round, homogeneous, hyperechogenic masses in the ventricles, and sometimes as multiple lesions in the ventricular free wall and interventricular septum.
There is a wide variation in the natural course and outcome of rhabdomyomas after birth [5,9]. From a histological point of view, rhabdomyomas are benign hamartomatous lesions, but in rare cases they may be lethal if heart failure and arrhythmias develop. Large masses may induce a decline of the right or left ventricular diastolic and systolic function, may mechanically interfere with cardiac valves or obstruct the LVOT and RVOT. Moreover, rhabdomyomas are potentially arrhythmogenic: supraventricular tachycardia causing foetal hydrops has been reported in the literature [6,10,11]. Some cases were found to have ventricular pre-excitation and incessant atrio-ventricular re-entry tachycardia immediately after birth [6,11].
A large retrospective study including 106 children with rhabdomyoma [6] showed that 10 patients (10%) had ventricular pre-excitation, and 6 (6%) had VT. The natural history of ventricular pre-excitation in these patients was mainly benign: 2 patients underwent successful radiofrequency catheter ablation and 8 patients were asymptomatic during follow-up. Among the 6 patients with VT, only 3 underwent surgical excision of a large tumour with complete disappearance of VT and one needed an implantable cardioverter defibrillator.
Surgery for resection of large cardiac tumours is not without risks. Operative management involves removal of the intracavitary portion of the tumour without complete excision of the entire lesion. Miyake et al. [6] reported three surgical deaths in 62 operated children (5%). All involved infants had large lesions for which transplant might have been the only alternative. Early or late appearance of post-operative ventricular aneurysms and systolic dysfunction are possible complications and deserve long-term attention with a close follow-up.
Recently, Martínez-García et al. [12] reported a significant size reduction of an inoperable giant left ventricular rhabdomyoma using everolimus. This may represent an alternative approach when surgery poses an unacceptable risk. Finally, the natural history of rhabdomyomatosis after birth may be characterized by neurological impairment due to tuberous sclerosis. This is an autosomal dominant Mendelian condition with a large variability of expression and its familial occurrence is reported in about 50% of cases. Our patient did not show a family history of rhabdomyomatosis or tuberous sclerosis, likely having a de novo gene mutation.
In conclusion, consistently with numerous reports, we observed a benign course of the disease in our patient with progressive and complete post-natal regression of rhabdomyomas. Owing to the favourable natural history of rhabdomyomas, most patients can be managed conservatively by echocardiography and 24-h Holter ECG monitoring. Patients with arrhythmias may be treated with antiarrhythmic medications and eventually by catheter ablation. Cardiac surgery is recommended only for patients with symptoms due to severe haemodynamic compromise, outflow tract obstruction, or life-threatening arrhythmias.
The authors confirm that written consent for submission and publication of this case report including images and associated text has been obtained from the patient in line with COPE guidance.
- Fesslova V, Villa L, Rizzuti T, Mastrangelo M, Mosca F (2004) Natural history and long-term outcome of cardiac rhabdomyomas detected prenatally. Prenat Diagn 24: 241-248. [Crossref]
- Smythe JF, Dyck JDS, Smallhorn JE, Freedom RM (1990) Natural history of cardiac rhabdomyoma in infancy and childhood. Am J Cardiol 66: 1247-1249. [Crossref]
- Holley DG, Martin GR, Brenner JI, Fyfe DA, Huhta JC, et al. (1995) Diagnosis and management of fetal cardiac tumors: a multicenter experience and review of published reports. J Am Coll Cardiol 26: 516-520. [Crossref]
- Harding CO, Pagon RA (1990) Incidence of tuberous sclerosis in patients with cardiac rhabdomyoma. Am J Med Genet 37: 443-446. [Crossref]
- Isaacs H Jr (2004) Fetal and neonatal cardiac tumors. Pediatr Cardiol 25: 252-273. [Crossref]
- Miyake CY, Del Nido PJ, Alexander ME, Cecchin F, Berul CI, et al. (2011) Cardiac tumors and associated arrhythmias in pediatric patients, with observations on surgical therapy for ventricular tachycardia. J Am Coll Cardiol 58: 1903-1909. [Crossref]
- Paladini D, Palmieri S, Russo MG, Pacileo G (1996) Cardiac multiple rhabdomyomatosis: prenatal diagnosis and natural history. Ultrasound Obstet Gynecol 7: 84-85. [Crossref]
- Nir A, Ekstein S, Nadjari M, Rass-Rotschild A, Rein AJ (2001) Rhabdomyoma in the fetus: Illustration of tumor growth during the second half of gestation. Pediatr Cardiol 22: 515-518. [Crossref]
- Tworetzky W, McElhinney DB, Margossian R, Moon-Grady AJ, Sallee D, et al. (2003) Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol 92: 487-489. [Crossref]
- Birnbaum SE, Mcgahan JP, Janos GG, Meyers M (1985) Fetal tachycardia and intramyocardial tumors. J Am Coll Cardiol 6: 1358-1361. [Crossref]
- Emmel M, Brockmeier K, Sreeram N (2004) Rhabdomyoma as accessory pathway: electrophysiologic and morphologic confirmation. Heart 90: 43. [Crossref]
- Martínez-García A, Michel-Macías C, Cordero-González G, Escamilla-Sánchez K, Aguinaga-Ríos M, et al. (2018) Giant left ventricular rhabdomyoma treated successfully with everolimus: case report and review of literature. Cardiol Young 28: 903-909. [Crossref]