Objectives: We have previously demonstrated association of the TNF Receptor Superfamily 6B gene (TNFRSF6B) encoding for DcR3, with Inflammatory Bowel Disease (IBD). Here we investigate the possible immuno-modulation mediated by DcR3 in EBV transformed cell lines from patient with and without risk variants in TNFRSF6B (rs2315008), in comparison with controls.
Methods: Expression of DcR3 and its kinetics were examined by immunoblot analysis in whole cell lysates from EBV transformed control and patient derived cell lines of different genotype background for rs2315008 (AA, AT, TT). Cell proliferation and cell death was measured by MMT assay. NF-kB kinetics was determined by immunoblot analysis of cytoplasm and nuclear extracts in a time course experiment. Caspase8, caspase9, caspase3, and Bcl2 expression were analyzed by western blots. DcR3 knockdown was performed using specific DcR3 siRNA. EBV cell lines from IBD patients harboring risk variants in the TNFRSF6B gene exhibit differential pattern of DcR3 expression and NF-kB activation and promote inflammation in Crohn’s Disease (CD).
Results: We have investigated the immuno-modulatory role of DcR3 in EBV transformed cell lines from patient with and without risk variants in TNFRSF6B. DcR3 induced rapid activation of nuclear factor kB (NF-kB) for all genotype states. However, EBV transformed cell lines derived from IBD patients harboring a risk variant in TNFRSF6B (A allele of the rs2315008 SNP) exhibit differential pattern of DcR3 expression and NF-kB kinetics in comparison with wild type. siRNA-mediated knockdown post 24hrs of nucleofection resulted in decreased DcR3 expression, increased cell death and decreased cell proliferation, effects that were also genotype-dependent. EBV cell lines from IBD patients harboring risk variants in the TNFRSF6B gene exhibit differential pattern of DcR3 expression and NF-kB activation and promote inflammation in Crohn’s disease by inhibiting FasL-induced apoptosis.
Conclusions: EBV cell lines from IBD patients harboring risk variants in the TNFRSF6B gene exhibit differential pattern of DcR3 expression and NF-kB activation and promote inflammation in Crohn’s disease (CD). We propose that pathogenic inflammation in CD may be in part be the result of non-canonical developmental signals impinging on a NF-kB signaling module with an altered homeostasis of IkB proteins.
Inflammatory Bowel Disease (IBD) collectively refers to 2 major chronic intestinal disorders of unknown etiology, Crohn Disease (CD) and Ulcerative Colitis (UC) and involves an overactive immune component in the intestinal mucosa [1,2]. Although both disease forms share some clinical and pathological features, the mechanisms underlying inflammation differ between CD and UC [3]. In a GWAS analysis carried out in a cohort of 1,011 individuals with pediatric-onset IBD and 4,250 matched controls, we identified and replicated a significantly associated, previously unreported locus on chromosomes 20q13 (rs2315008 [T] and rs4809330 [A]; P = 6.30 × 10−8 and 6.95 × 10−8, respectively; odds ratio (OR) = 0.74 for both) located close to the TNFRSF6B gene [4]. Association at this locus continued to strengthen in the recent meta-analyses of Crohn’s disease [5] after confirming replication of the locus. The study also looked for difference in serum DCR3 concentration between individuals with IBD and controls and, within the IBD group, between those with and without the identified at-risk variants captured by the TNFRSF6B tagging SNPs and found that mean serum DCR3 concentration was increased approximately 50-fold in individuals with IBD and was highest in those with IBD carrying the minor allelic variant, which is protective of IBD. The individuals with IBD carrying the protective allele did not otherwise differ based upon age, gender, clinical disease activity or concurrent medications from those IBD patients carrying the risk allele, suggesting that the difference in serum DCR3 concentration between the two groups (~3-fold) was due to the observed genotypic variation at 20q13 [4]. On the basis of differences in serum DCR3 concentration between controls and individuals with IBD with and without the identified SNPs, and its known biologic function, TNFRSF6B is the most plausible candidate within the 20q13 locus, as the expression of other genes at this locus was unaltered. TNFRSF6B encodes for Decoy Receptor 3 (DcR3) protein, which binds to cytokines of the tumor necrosis factor superfamily, namely TL1A (TNFSF15), LIGHT (TNFSF14) and Fas ligand (FASLG) and blocks their ability to stimulate their receptors [5,6]. To better characterize the role of DcR3 variants in IBD, we sequenced the exons of TNFRSF6B in a large number of Caucasian pediatric IBD cases and healthy controls. We uncovered several missense variants at the TNFRSF6B locus, affecting secretion of DcR3 from cultured cells that were significantly enriched in the IBD cases [7]. Since these cytokines are pro-inflammatory, deficiency of DcR3 expression, secretion, or ligand-binding leads to unopposed inflammatory signals and exacerbation of IBD and suggests that DcR3 may be less effective in downregulating ligands that provoke inflammation in the IBD cases [8]. Thus, DCR3 has multiple complex roles within the innate and adaptive immune system, which may result in a net pro- or anti-inflammatory effect based upon the precise context and further modified by specific sequence variants [7].
The role of NF-kB in the pathogenesis of IBD has been examined in recent studies. Colon biopsies from IBD patients with active disease show increased levels of NF-kB p65 protein, which correlates with the severity of intestinal inflammation [9]. Aberrant activation of the transcription factor NF-kB controls the expression of many genes of inflammatory cytokines involved in the pathogenesis of IBD [10]. Here we investigate the immuno-modulatory role of DcR3 in EBV transformed pediatric control and patient-derived cell lines with and without risk variants in TNFRSF6B, best captured by the tagging SNP, rs2315008.
Because DcR3 lacks a transmembrane sequence and is a soluble protein, we used an immunoblot assay to determine the expression of DcR3 in whole cell lysates and culture supernatant from control and patient-derived Immortalized cell lines with and without risk variants in the TNF Receptor Superfamily 6B gene using a mouse monoclonal antibody to DcR3. We used a non-secretor cell line as a baseline control for the development of the assay model [7]. The ability of DcR3 to induce IkBα degradation was determined by immunoblot analysis. Briefly, 1×106 cells derived from control and IBD patients EBV transformed cell lines harboring risk variants in TNFRSF6B (A allele) were used in a time course experiment to evaluate the kinetics of IkBα degradation for time points ranging from 0 min to 60 min. Cells were then lysed in NuPAGE LDS sample buffer (Invitrogen Life Technologies) and boiled for 5 min before loading. A total of 10ug of protein per sample was separated on 4-12% Bis-Tris density gradient gels in MOPS SDS running buffer and transferred to nitrocellulose membrane membranes (Invitrogen Life Technologies), which after blocking with 3% BSA and 0.1% Tween 20 were incubated with rabbit polyclonal anti- IkBα C-21 (Santa Cruz Bio- technology). Bound Ab was detected using HRP-conjugated donkey anti-rabbit (Amersham Biosciences) and ECL detection system (Amersham Biosciences). Where specified, membranes were stripped in 0.2 M glycine (pH 2.5), 0.05% Tween 20, and 140 mM NaCl in TBS at 50°C for 30 min, blocked with 3% BSA, and reprobed with mouse anti-β-actin monoclonal Ab (Santa Cruz Bio- technology). The effect of DcR3 on cell proliferation was measured by the colorimetric MTT assay using an MTT (Microtiter-tetrazolium, Sigma, USA) based assay. Briefly, the cells (5,000/ml) were incubated in triplicate in a 96-well plate (Costar, Cambridge, MA, USA) in a final volume of 0.2 ml for the indicated times. Thereafter, 20 μl of MTT solution (5 g/L) was added to each well and then incubated for 12 h. After centrifugation, the supernatant was removed from each well. The colored formazan crystal produced from MTT was dissolved in 0.15 ml of DMSO and then the Optical Density (OD) value was measured on a microplate reader. Each experiment was performed in duplicate and repeated five times.
NF-kB is one of the key regulators in the immunological setting of IBD and therefore appears as a very promising target for therapeutic intervention in IBD. We next wanted to determine whether having different combinations of risk variant does affect DcR3 capability of inducing NF-kB activation differentially and impacting its functions. NF-kB kinetics was determined by immunoblot analysis of cytoplasm and nuclear extracts in a time course experiment. Nuclear extracts were prepared by washing cells twice with 1 ml of ice-cold PBS and resuspended in 400ul of ice-cold lysis buffer containing 1 M HEPES, 0.5M EDTA, 0.1M EGTA, 2M KCl, 0.1M DTT, mix of protease inhibitors at 5ug/ml (Roche), and 10% Nonidet P-40 and incubated for 15 min on ice with occasional vortexing to obtain complete cell lysis and release of nuclei. Tubes were centrifuged at 13,400 xg for 1 min, supernatant (cytoplasmic extract) was collected and remaining nuclei were resuspended in 25ul of ice-cold nuclear extraction buffer containing 1 M HEPES, 5 M NaCl, 0.5 M EDTA, 0.1 M DTT, and mixture of protease inhibitors at 5ug/ml (Roche), incubated for 30 min on ice and centrifuged at 13,400 x g for 5 min. Supernatant containing the soluble nuclear proteins was aliquoted in prechilled tubes, snap-frozen in liquid nitrogen and stored at -80°C until use. Equal protein amounts of the extracts (10ug) as determined using detergent compatible protein assay (Bio-Rad) were used in experiments. Considering that apoptosis plays a central role in regulation of tissue homeostasis, the imbalance between cell death and proliferation in favour of cell survival could result in chronic intestinal inflammation. We next investigated the differential effect of having risk allele variant of DcR3 on the extrinsic and intrinsic cell apoptosis pathway and cell survival. Caspases were determined by western blot analysis. Aliquots (10 μg of protein) of whole cell lysates were separated on 4-12% Bis-Tris gels, blotted onto a nitrocellulose membrane and probed with antibodies against Caspase-8, Caspase-3, Caspase-9 and Bcl-2 (Sigma, USA). Membranes were washed with 0.05% (vol/vol) Tween 20 in PBS (pH 7.6) and incubated with 1: 10,000 dilution of horseradish peroxides-conjugated secondary Abs for 60 min at room temperature. Bound Ab was detected using ECL detection system (Amersham Biosciences). Where specified, membranes were stripped in 0.2 M glycine (pH 2.5), 0.05% Tween 20, and 140 mM NaCl in TBS at 50°C for 30 min, blocked with 3% BSA, and reprobed with mouse anti-b-actin monoclonal Ab (Santa Cruz Bio- technology). Although NF-kB is one of the key regulators in the immunological setting of IBD and therefore appears as a very promising target for therapeutic intervention in IBD, it is nevertheless important to remember that NF-kB is also involved in normal cell physiology. We next investigated the effect of having risk allele variant of DcR3 in the context of NF-kB modulation. The NF-kB family of transcription factors contains five members, RelA (p65), RelB, c-Rel, NF-kB1 (p50), and NF-kB2 (p52). The existence of diversity among these proteins has raised the possibility that specific functions can be induced by particular hetero-dimers or homo-dimers in response to distinct stimuli. The most typical are hetero-dimers consisting of p65 (RelA) and p50 or c-Rel. Because little is known about the specific paradigm of NF-kB activation and the role that individual NF-kB family members may play in pathogenesis of human IBD, we wanted to understand the basic physiology of activation and expression pattern of NF-kB complexes in non-secretors, control and patient-derived EBV transformed cell lines.
Previous studies have demonstrated that DcR3 acts as a decoy receptor and neutralizes the FasL-mediated apoptotic signal [11]. The expression of DcR3 was reduced using siRNAs in the EBV transformed cell lines derived from control and IBD patients harboring risk variants in TNFRSF6B (A allele). Briefly, 3X106 cells were transfected with 100nM DcR3 siRNA duplexes or with DcR3 nonsilence control using Amaxa Nucleofector Kit and and program T-20. The siRNA-transfected cells were incubated for 48 h after nucleofection and the degree of knockdown relative to introduction of control siRNA was confirmed by western blot using DcR3 mouse monoclonal antibody (Abgent) on whole cell lysates. The 21-mer siRNAs were synthesized by DHARMACON. The DcR3 siRNA sequences were as follows: sense sequence, 5’-GCC AGG CUC UUC CUC CCA UdTdT-3’; antisense sequence, 5’- AUG GGA GGA AGA GCC UGG CdTdT-3’. The nonsilence control siRNA sequences were as follows: sense sequence, 5’-GCC CGC UUU CCC UCA GCA UdTdT-3’; antisense sequence, 5’-AUG CUG AGG GAA AGC GGG C-3.
As shown in Figure 1, control and patient homozygous for risk allele 1 and 2 (1 stands for A or T and 2 stands for G or C) showed similar levels of endogenous DcR3 protein expression in whole cell lysates and supernatant. In comparison to control, the heterozygous patient (1/2) showed increased DcR3 protein levels in both whole cell lysates and supernatant. However, the non-secretor showed low endogenous DcR3 levels of protein in whole cell lysates with no detectable levels in supernatant, highlighting the secretion defect in human-derived EBV cells from IBD patients and controls, we described previously in transfected 293T cells [7].
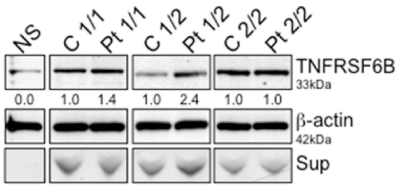
Figure 1. DcR3 western blot analysis. The whole cell lysate of non-secretor, control and patient derived EBV cells was analyzed for DcR3 expression. 1 stands for A or T. 2 stands for G or C. Blots were stripped and reprobed for β-actin as a loading control; numbers beneath each lane represent densitometric ratios of candidate protein normalized to the loading control
Next, we initially evaluated the kinetics of IkBα degradation in these cell lines for times ranging from 0 min to 60 min without any stimuli. As shown in Figure 2, in comparison to controls patient homozygous for risk variant allele 1 showed early maximum IkBα degradation by 30 min. However, the patient with risk allele variant homozygous (2/2) and heterozygous (1/2) showed extended IkBα degradation up to 60 mins. To further confirm that this activity was restricted specifically to patient cells, control and patient cells from each group were subjected to treatment of RPMI medium, control supernatant, DcR3-containing supernatant and vice versa for 60 min. In all the cases short-term treatment of control cells with DcR3 supernatants from patients resulted in a substantial degradation of IkBα, which indicated NF-kB activation (Figure 3).
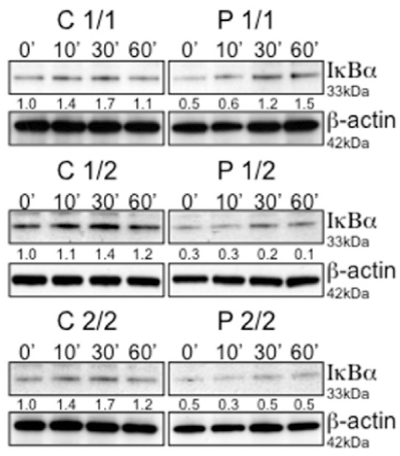
Figure 2. Activation and kinetics of NF-kB was monitored by immunoblot analysis of IkBα in a time course experiment. Lysates of 106 cells were separated by SDS-PAGE and evaluated for IkBα degradation in a time course experiment. Blots were stripped and reprobed for β-actin as a loading control. Numbers beneath each lane represent densitometric ratios of candidate protein normalized to the 0-minute time point
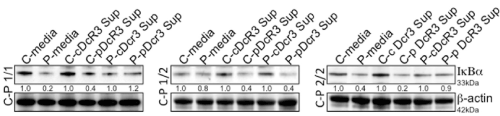
Figure 3. Decoy receptor 3 (DcR3) induces the activation of NF-kB. Activation of NF-kB was monitored by immunoblot analysis of IkBαin control and patient cells with three different combinations of risk variants (1/1, 1/2 and 2/2). Cells were incubated with RPMI1640 medium (DMEM), control supernatant, and DcR3-containing supernatant for 30 min. Blots were stripped and reprobed for b-actin as a loading control; numbers beneath each lane represent densitometric ratios of candidate protein normalized to the loading control
We next investigated the differential effect of having risk allele variant on cell proliferation. Control and patient cells with three different combinations of risk variants (1/1, 1/2 and 2/2) and non-secretor cells were subjected to MTT proliferation assay. The patient cells with 1/2 heterozygous and 2/2 homozygous risk alleles exhibited maximum proliferation in comparison to controls. Non-secretors exhibited lower rate of proliferation (Figure 4). These data suggest that having different combinations of risk variant does affect functions mediated by DcR3 differentially.
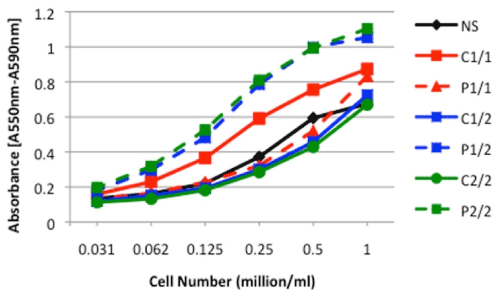
Figure 4. MTT analysis. The cell proliferation was measured using an MTT (Microtiter-tetrazolium) based assay in non-secretors, control and patient cells with three different combinations of risk variants (1/1, 1/2 and 2/2). The data is representative of five separate experiments
Western blot analysis was performed for protein expressions of caspase-8/9/3 and anti-apoptotic Bcl-2 on whole cell lysates of non-secretors, control and patient EBV transformed cells in a time course experiment, ranging from 0 min to 60 min (Figure 5A). Patient cells with homozygous risk allele 1 exhibit no difference in caspase-8, 9 and 3 levels in comparison with control cells; however, they exhibit high anti-apoptotic Bcl2 protein levels. The patient EBV cells with heterozygous risk allele (1/2) exhibits high levels of capase-9 and 3 and high levels of anti-apoptotic Bcl2 protein levels in comparison with control cells. The patient EBV cells harboring homozygous risk allele 2 showed decreased levels of caspase 8,9,3 and low levels of anti-apoptotic Bcl2 protein in comparison with control cells. However, the non-secretors showed only caspase 8 and 9 with no detectable levels of caspase 3 and low levels of anti-apoptotic Bcl2 over the time course as depicted in bar graph (Figure 5B). These data suggest that having different combinations of DcR3 risk variant differentially regulates and activates cell death and survival functions.
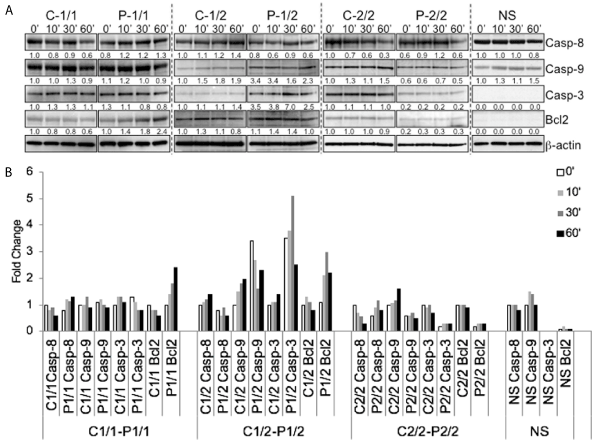
Figure 5. Detection of Caspase-8, Caspase-3, Caspase-9 and Bcl-2. (A) Lysates from 106 cells of non-secretors, control and patient cells with three different combinations of risk variants (1/1, 1/2 and 2/2) were separated by SDS-PAGE and evaluated for Caspase-8, Caspase-3, Caspase-9 and Bcl-2 in a time course experiment. Blots were stripped and reprobed for β-actin as a loading control. Numbers beneath each lane represent densitometric ratios of candidate protein normalized to the 0-minute time point and loading control. (B) bar graph for expression levels and fold change over time of caspase-8, caspase-3, caspase-9 and Bcl-2
To determine whether enhanced expression and localization of NF-kB components is also detectable in the pediatric IBD patients, the cytoplasm as well as nuclear extracts was prepared from non-secretors, control and patient transformed EBV cell lines. The presence and precise nuclear translocation of different NF-kB family members was determined by western blot in a time course experiment (Figure 6A & 6B). β-actin is included as a control for cytoplasmic extracts and also confirms the actin-poor nuclear fraction. Patient cells with homozygous risk allele 1 exhibit increased nuclear localization of c-Rel, p52 (non-classical component) and similar levels of classical heterodimers p65 and p50 in comparison with control cells. Another important finding is the existence of members of both classical and the non-classical NF-kB pathway in the nucleus of patient EBV cells with heterozygous risk allele (1/2). The patient EBV cells with heterozygous risk allele (1/2) exhibit relatively high nuclear levels of p50, c-Rel, RelB and p52 and similar levels of p65 in comparison with control cells. The patient EBV cells harboring homozygous risk allele 2 show high levels of p50 and similar levels of p65 and p52 in comparison with control cells. However, the non-secretors showed only the members of Classical NF-kB pathway in the nucleus. In addition, p50 is preferentially concentrated in the nucleus of all resting control and patient EBV cells irrespective of their allelic background and would be consistent with the existence of nuclear p50 homodimers that have been previously described to serve an inhibitory role (Moynagh). These data suggest that having different combinations of DcR3 risk variant differentially regulates expression and localization of NF-kB family members.
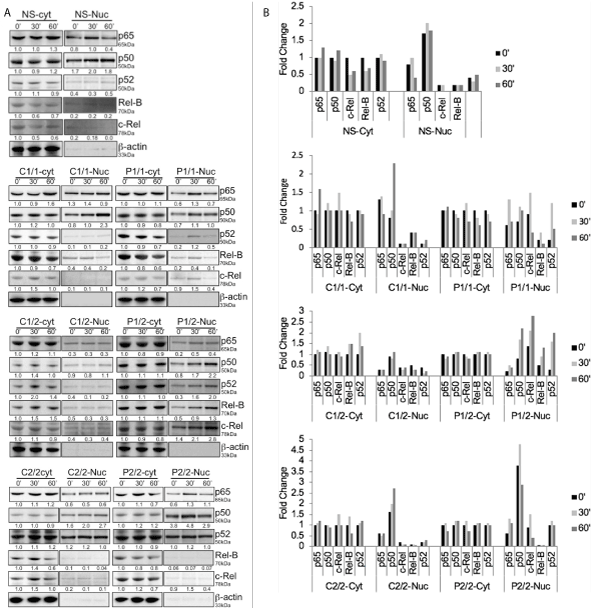
Figure 6. Cytoplasmic and nuclear expression levels of specific NF-kB members. (A) Cytoplasmic and nuclear levels of specific NF-kB members was monitored by immunoblot analysis. Blots were stripped and reprobed for β-actin as a loading control in cytoplasmic extracts. No β-actin in nuclear extracts represents the purity of nuclear extracts. Numbers beneath each lane represent densitometric ratios of candidate protein normalized to the 0-minute time point and loading control. (B) Bar graph represents fold change in cytoplasmic and nuclear expression levels of specific NF-kB members in non-secretors, control and patient cells with three different combinations of risk variants (1/1, 1/2 and 2/2) monitored by immunoblot analysis in a time course experiment
Since the patient EBV cells with heterozygous risk allele (1/2) exhibit more aggressive inflammatory marker upregulation in our experimental model, we next examined whether these EBV cells are benefitted from knockdown of DcR3 expression. Figure 7A & 7B shows the decrease in DcR3 expression post nucleofection of P (1/2) EBV cells with control siRNA and DcR3 siRNA. Post 24, 48 and 72 hours of nucleofection, cells were harvested and whole cell lysates were evaluated for DcR3 expression by immunoblot analysis. Blot shows reduced DcR3 levels first seen at 24h and maintained for at least 48h. Nucleofection had no adverse effect on cell viability (data not shown). In addition, cells nucleofected with control and DcR3 siRNA post 24h were treated with sFasL and analyzed for cell death using MTT Proliferation assay. Cells receiving DcR3 siRNA treated with sFasL showed decrease in cell growth in comparison to cells receiving the CsiRNA and sFasL (Figure 7C). These results suggest that siRNA knockdown of DcR3 expression rescues the cytotoxic effect of FasL.
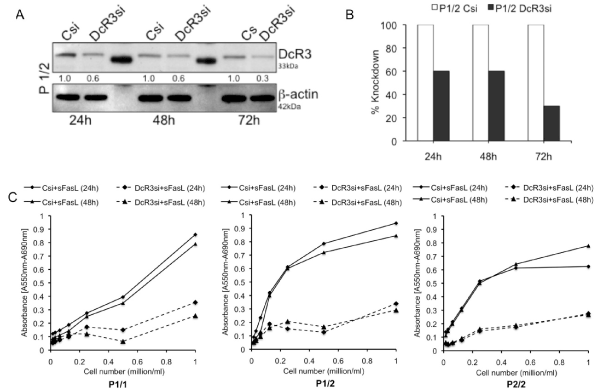
Figure 7. siRNA knockdown of DcR3 expression. (A) & (B) shows the decrease in DcR3 expression post nucleofection of P (1/2) EBV cells with control siRNA and DcR3 siRNA. (C) Represents effect of DcR3 knockdown on cell proliferation measured by MTT assay in patient cells with three different combinations of risk variants (1/1, 1/2 and 2/2)
In this study, we report that patients harboring risk variants in the TNFRSF6B gene exhibit differential pattern of DcR3 expression and NF-kB activation. Control and patient homozygous for risk allele 1 or 2 (1 stand for A or T and 2 stands for G or C) show similar levels of endogenous DcR3 protein expression in whole cell lysates and supernatant. In comparison with controls, cells from the heterozygous patients (1/2) show increased DcR3 protein levels in both whole cell lysates and supernatant. Thus, the risk variants carrier stage has an impact on the levels of DcR3. We also obtained evidence that different combinations of risk variants affect DcR3 capability of inducing NF-kB activation differentially and this has functional impact. In comparison with controls, cells from patients homozygous for risk variant allele 1 show early maximum IkBα degradation by 30 min. However, patients who are either homozygous (2/2) or heterozygous (1/2) for the risk allele showed extended IkBα degradation up to 60 mins and this DcR3 mediated rapid NF-kB activation activity was restricted specifically to patient cells harboring the risk variants.
The above results prompted us to investigate the effect of risk alleles at the DcR3 locus on the extrinsic and intrinsic cell apoptosis pathway and cell survival as a functional correlate. Binding of CD95L to CD95 initiates the extrinsic apoptosis pathway. CD95, FADD and caspase 8 form the DISC to activate pro-caspase 8. Caspase 8 then activates caspase 3 by cleavage resulting in apoptosis. The intrinsic pathway triggered by stress stimuli is initiated by activation of Bim. Bim inhibits Bcl-2/BcL-xL, which leads to activation of Bak and bax. These molecules trigger the release of cytochrome c from mitochondria, which in turn is recruited into the apoptosome. Caspase 9 is activated in the apoptosome and finally activates caspase3 and induces apoptosis. Patient cells with homozygous risk allele 1 exhibit no difference in caspase-8, 9 and 3 levels in comparison with control cells, however, they exhibit high anti-apoptotic Bcl2 protein levels. The patient EBV cells with heterozygous risk alleles (1/2) exhibit high levels of capase-9 and 3 and high levels of anti-apoptotic Bcl2 protein levels in comparison with control cells. The patient EBV cells harboring homozygous risk allele 2 showed decreased levels of caspase 8,9,3 and low levels of anti-apoptotic Bcl2 protein in comparison to control cells. In contrast, the non-secretors showed only caspase 8 and 9 with no detectable levels of caspase 3 and low levels of anti-apoptotic Bcl2 over time course. These data suggest that different combinations of DcR3 risk variants differentially regulate and activate cell death and survival functions.
Because little is known about the specific paradigm and considering the different cell-type specific effects which are mediated by NF-kB, we wanted to understand the basic physiology of activation and expression pattern of NF-kB complexes in non-secretors, including both controls and patient-derived EBV transformed cells. Patient cells with homozygous risk allele 1 showed increased nuclear localization of c-Rel, p52 (non-classical component) and similar levels of classical heterodimers p65 and p50 in comparison with control cells.
Another important finding is the existence of both classical and non-classical NF-kB pathway members in the nucleus of patient EBV cells with heterozygous risk allele (1/2). The patient EBV cells with heterozygous risk allele (1/2) exhibit relatively high nuclear levels of p50, c-Rel, RelB and p52 and similar levels of p65 in comparison with control cells. The patient EBV cells harboring homozygous risk allele 2 show high levels of p50 and similar levels of p65 and p52 in comparison with control cells. Though, the non-secretors exhibit deficiency of DcR3 expression, secretion, or ligand-binding, presence of members of Classical NF-kB pathway in the nucleus may lead to unopposed inflammatory signals and exacerbation of IBD and suggests that DcR3 may be less effective in downregulating ligands that provoke inflammation in the IBD cases [7]. In addition, p50 is preferentially concentrated in the nucleus of all resting control and patient EBV cells irrespective of their allelic background and would be consistent with the existence of nuclear p50 homodimers that have been previously described to serve an inhibitory role. Thus, increased processing of p105 and rapid degradation of IκBα by immunoproteasomes in CD patients may be responsible for enhanced expression of inflammatory genes regulated by p50/c-Rel and p50/p65 heterodimers. While previous studies suggest that NF-kB p65 is involved in the pathogenesis of CD, our data show that the abundance and mechanism of induction differs between individuals with different combinations of DcR3 risk variants. Most NF-kB inhibitors on the market and in development target only the better-known NF-kappa B canonical pathway. Our results indicate that the DcR3 is capable of inducing both canonical and non-canonical NF-kB pathways and the lesser-known non-canonical pathway actually may play a more important role in disease progression and pathogenesis. The five members of the NF-kB family of proteins - also called REL proteins can form multiple dimers, including at least five that have known transcriptional activity. Their activation can increase the expression of pro-growth and anti-apoptotic genes in cancer and increase the expression of pro-inflammatory genes in autoimmune diseases like IBD. The dimers are normally kept inactive by NF-KBIa(nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha) (12).
Many upstream signaling processes induce their activation through only one of the two pathways: the canonical or non-canonical (13). Most efforts to develop drugs that block NF-kB activation have focused on the canonical pathway because its targets - such as tumor necrosis factor (TNF) alpha - are better understood and easier to inhibit. But growing body of evidence shows the non-canonical pathway may play a more important role in disease. In 2007, researchers at Profectus Biosciences Inc. who were studying a connection between NF-kB and CC chemokine receptor 5 (CCR5; CD195) in HIV infection identified small molecule REL inhibitors. While Profectus’ compounds had antiviral properties, the company thought cancer presented a greater unmet medical need and a better prospect for success. To maintain its infectious disease focus, Profectus spun out the REL inhibitor program into Rel-MD in 2011. The lead REL inhibitor, PBS-1086 (REM-1086), inhibits all five REL proteins and thus blocks both the canonical and non-canonical pathways. The five REL proteins are nuclear factor of kappa light polypeptide gene enhancer in B cells 1 (NFKB1; p105; p50); nuclear factor of kappa light polypeptide gene enhancer in B cells 2 p49/p100 (NFKB2; p52); v-rel reticuloendotheliosis viral oncogene homolog A (RELA; p65); v-rel reticuloendotheliosis viral oncogene homolog avian (CREL; REL); and v-rel reticuloendotheliosis viral oncogene homolog B (RELB). The canonical pathway activates dimers formed by p50 and either RELA or C-REL, while the non-canonical pathway activates dimers formed by p52 and RELA, RELB or C-REL (13). Rel-MD chose Multiple Myeloma (MM) as the lead indication based on a pair of studies reported by the Dana-Farber Cancer Institute. One study showed that an off-target effect of Velcade bortezomib was to induce up-regulation of the canonical pathway in MM cell lines and tumors (14). The other study showed that inhibiting both NF-kB activation pathways in MM cell lines was more effective than targeting just one (15). Thus, the non-canonical pathway may play a more important role in the development of new therapies in the future.
Taken together, we have investigated the immuno-modulatory role of DcR3 in EBV transformed cell lines from patient with and without risk variants in TNFRSF6B. DcR3 induced rapid activation of nuclear factor kB (NF-kB) for all genotype states. However, EBV transformed cell lines derived from IBD patients harboring a risk variant in TNFRSF6B (A allele of the rs2315008 SNP) exhibit differential pattern of DcR3 expression and NF-kB kinetics in comparison with wild type. siRNA-mediated knockdown post 24hrs of nucleofection resulted in decreased DcR3 expression, increased cell death and decreased cell proliferation, effects that were also genotype-dependent. EBV cell lines from IBD patients harboring risk variants in the TNFRSF6B gene exhibit differential pattern of DcR3 expression and NF-kB activation and promote inflammation in Crohn’s disease by inhibiting FasL-induced apoptosis. Our results offer first experimental evidence of involvement of non-canonical NF-kB signaling in pathogenesis of CD. We propose that pathogenic inflammation in CD may be in part be the result of non-canonical developmental signals impinging on a NF-kB signaling module with an altered homeostasis of IkB proteins. We anticipate that our findings will be extended to establish the non-canonical NF-kB pathway as a key player in the pathogenesis of IBD and may lead to the development of new therapies in the future.
We thank the patients and control subjects for their invaluable contribution to the study. Funding: This work is supported by an Institutional Development Fund from The Children’s Hospital of Philadelphia and a Research Grant from the Crohn’s and Colitis Foundation of America.
Study concept and design: R.P., R.N.B., H.H. Technical or material support: E.C.F., C.E.K., R.M.C., R.P. Drafting of the manuscript: R.P., H.H. Obtained funding: R.N.B., H.H. Study supervision: R.N.B., H.H.
The authors have no competing interests to declare.
- Shih Q, Targan R (2009) Insights into IBD Pathogenesis. Curr Gastroenterol Rep 11: 473-80. [Crossref]
- Kaser A, Zeissig S, Blumberg RS (2010) Inflammatory bowel disease. Annu Rev Immunol 28: 573-621. [Crossref]
- Denson LA (2013) The Role of the Innate and Adaptive Immune System in Pediatric Inflammatory Bowel Disease. Inflamm Bowel Dis 19: 2011-2020. [Crossref]
- Kugathasan S, Baldassano R, Bradfield J, Sleiman PM, Imielinski M, et al. (2008) Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat Genet 40: 1211-1215. [Crossref]
- Franke A, McGovern D, Barrett J, Wang K, Radford-Smith GL, et al. (2010) Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 42: 1118-1125. [Crossref]
- Lin W, Hsieh S (2011) Decoy receptor 3: a pleiotropic immunomodulator and biomarker for inflammatory diseases, autoimmune diseases and cancer. Biochem Pharmacol 81: 838-847. [Crossref]
- Kim S, Fotiadu A, Kotoula V (2005) Increased expression of soluble decoy receptor 3 in acutely inflamed intestinal epithelia. Clin Immunol 115: 286-294. [Crossref]
- Christopher J, Zhi W, Saarene P et al. (2013) Targeted Resequencing Identifies Secretion-Defective Variants of Decoy Rceptor 3 in Pediatric-Onset Inflammatory Bowel Disease.
- Neurath M, Pettersson S, Meyer zum Büschenfelde K, Strober W (1996) Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med 2: 998-1004. [Crossref]
- Neurath M, Fuss I, Schürmann G, Pettersson S, Arnold K, et al. (1998) Cytokine gene transcription by NF-kappa B family members in patients with inflammatory bowel disease. Ann N Y Acad Sci 859: 149-159. [Crossref]
- Wortinger M, Foley J, Larocque P, Witcher DR, Lahn M, et al. (2003) Fas ligand-induced murine pulmonary inflammation is reduced by a stable decoy receptor 3 analogue. Immunology 110: 225-233. [Crossref]
- Ghosh S, Baltimore D [1990] Activation in vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B. Nature 344: 678-682. [Crossref]
- Hayden M, Ghosh S (20014) Signaling to NF-kappaB. Genes Dev 18: 2195-2224. [Crossref]
- Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, et al. (2009) Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 114: 1046-1052. [Crossref]
- Fabre C, Mimura N, Bobb K, Kong SY, Gorgun G, et al. Dual inhibition of canonical and noncanonical NF-κB pathways demonstrates significant antitumor activities in multiple myeloma. Clin Cancer Res 18: 4669-4681. [Crossref]