Aim: Gestational diabetes mellitus (GDM) is a complex metabolic disorder with largely unknown etiology. GDM is associated with fetal morbidity and mortality. This study aims to explore the novel players involved in the pathophysiology of GDM.
Materials and methods: The raw gene expression profile (ID: GSE49524) was downloaded from the Gene Expression Omnibus database. The differentially expressed genes were identified (DEGs, criteria: p-value<0.05 and log2 FC>0.1). Gene pairs with a combined score >0.9 were utilized to construct a protein-protein interaction (PPI) network. These DEGs in PPI were then compared with the known disease genes downloaded from OMIM and Gene Cards to find candidate GDM related genes. Furthermore, pathway and functional enrichment analyses were performed through (criterion: p-value<0.05).
Results: A total of 571 DEGs were identified of which only 170 DEGs with a combined score >0.9 were utilized to construct a PPI network. Of these 170 DEGs, 96 genes were up, while 74 genes were down-regulated. Moreover, 8 known disease genes of GDM were obtained. Genes in the PPI network were then significantly enriched in several functions separately for up and down-regulated genes while 6 pathways in combined. Based on PPI network and functional consistency, 8 genes namely IL13RA1, HSPA5, PLAU, TGFA, PLAT, BID, SP1, and NUP50 were identified as candidate GDM related genes. Out of these, PLAT and HSPA5 were found to be least reported in GDM and may serve as novel candidate GDM related genes. However, further studies are required to validate these results.
Conclusions: IL13RA1, HSPA5, PLAU, TGFA, PLAT, BID, SP1, and NUP50 were identified as candidate GDM related genes. Of these, HSPA5 and PLAT can serve as novel biomarker of GDM.
- Total 571 DEGs were identified with 302 up-regulated and 269 down-regulated genes.
- Based on functional consistency, IL13RA1, HSPA5, PLAU, TGFA, PLAT, BID, SP1, and NUP50 were identified as candidate GDM related genes.
- These genes were enriched in total 6 pathways.
- GDM affects prenatal growth by altering umbilical cord’s gene expression.
- This study provides insight into the mechanism involved in prenatal morbidity associated with GDM.
gestational diabetes mellitus, protein-protein interaction, differentially expressed genes, gene ontology
Gestational diabetes mellitus (GDM) is the most common medical complication of pregnancy. It is a type of diabetes that arises during the gestational period and can be formally defined as glucose intolerance with onset or first recognition during pregnancy, affecting around 2-5% of all pregnancies [1]. In the high-risk population, it may affect up to 16% of total pregnant women [2]. During the past few years, there is an exponential rise in its prevalence worldwide. The prevalence rates for GDM are higher for African, Hispanic, Indian, and Asian women than for Caucasian women [3,4]. Recently, the prevalence of GDM has increased by 2-3 folds, ranging from 8.9-53.4% [5]. This rise in GDM incidence is of prime concern as it deteriorates not only maternal health but also fetal health.
Gestational diabetes is a metabolic disorder in which altered glucose metabolism affects the maternal as well as fetal molecular, cellular, and physiological health. During pregnancy, insulin resistance occurs to ensure proper nourishment of growing foetus, in order to compensate this insulin demands increases, and failure of β-cell to compensate this extra demand of insulin causes hyperglycemic state leading to GDM [6]. GDM is associated with adverse maternal and neonatal outcomes which can be short as well as long term. Short term maternal complications are hypertension, preeclampsia, and an increased risk of the cesarean section while in long term consequences they may develop diabetes later in life [7]. Neonatal complications include overweight at delivery, hypoglycemia, macrosomia, cardiac dysfunction, and congenital malformations [8]. Recently, it has been shown that epigenetic reprogramming of gene expression in fetus genome is associated with diabetes [9], possibly due to hyperglycemia that triggers specific molecular effects during prenatal growth.
Despite its increasing incidence and a wide spectrum of maternal and fetal pathologies, no specific genetic factors associated with GDM have been identified yet. In the present study, we have tried to identify the novel candidate gene of GDM. To achieve the target, gene expression datasets with ID GSE49524 was used and analyzed to identify the differentially expressed genes (DEGs) between non-diabetic and diabetic mothers. The dataset contains the transcriptome of human umbilical vein endothelial cells (HUVEC) obtained from the vein of umbilical cords collected from GD patients and compared to control cells obtained from healthy donors. Based on the combined score, the protein-protein interaction network was constructed. DEGs in PPI was then compared with known disease genes to identify novel candidate GDM related genes. Functional and pathway enrichment analysis was performed for DEGs along with candidate GDM gene interacting in the network to identify the processes and pathways in which they are involved. Our results indicate the role of many novel players in determining the pathophysiology of GDM and hence altering the embryo development via epigenetic reprogramming.
Microarray data
The raw gene expression profile datasets (ID: GSE49524) deposited by Leoni G, et al. [10] were downloaded from the National Center of Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database [11]. The samples for this data were cultured human primary endothelial cells (HUVECs) from umbilical cords of 3 Gestational Diabetes mothers and 3 non-diabetic mothers. The dataset contains 6 chips derived from a study using the GPL7020 NuGO array (human) NuGO_Hs1a520180 platform. Umbilical cords were collected from 3 Caucasian GDM patients and 3 non-diabetic mothers at 39 to 41 weeks immediately after delivery. Both groups were compared for anthropometric and biochemical parameters. Maternal age (GDM 35.6 ± 4.0 and control 34.4 ± 4.5), pre-gestational weight (GDM 62.1 ± 13.7 and control 67.9 ± 29.3, kg), height (GDM 1.62 ± 0.09 and control 1.66 ± 0.04, mt), pre-gestational body mass Index (GDM 25.5 ± 2.4 and control 22.5 ± 3.7) and fasting glycemia (GDM 5.16 ± 0.48 and control 4.33 ± 0.66, mM) were the parameters which were analyzed, data have not been shown for other biochemical parameters. All procedures were in agreement with the ethical standards of the Institutional Committee on Human Experimentation.
Data preprocessing and DEGs screening
The preprocessing of downloaded raw gene expression datasets were performed. First, to get the gene expression value, expression values of probes were averaged corresponding to a specific gene. Then, BiGGEsTs [12] software analysis was performed to select exclusively up-regulated and down-regulated genes. Furthermore, official gene symbols were extracted for each corresponding probe-level symbols by using GEO2R [13]. All DEGs selected have p values <0.05 and threshold logFC values >0.1 for up and <-0.1 for down genes.
Principal component analysis and heat map generation
Principal component analysis (PCA) was performed for significant DEGs only with the help of online ClustVis tool [14] as it supports maximum file size up to 2 MB and hence it was not possible to prepare the PCA plot for total gene expression profile. Singular value decomposition (SVD) with imputation was used to calculate the principal component. Similarly, heatmap was generated for DEGs using same online ClustVis tool, both rows and columns were clustered using correlation distance and average linkage.
Construction of PPI network and sub-network mining
Identified DEGs were uploaded to STRING v 10.5 [15] which gives a combined score (CS) for protein-protein interaction among gene pairs. Combined score (CS) >0.9 was considered as criteria for selection. Protein-protein Interaction (PPI) network was created for DEGs with CS >0.9 by using Cytoscape v 3.2.1 [16] followed by sub-network mining. Average shortest path length and edge betweenness were taken as criteria for network construction.
Functional enrichment analysis
The database for annotation, visualization, and integrated discovery (DAVID v 6.7) [17] was used for Gene ontology (GO) enrichment analysis including biological process (BP), molecular function (MF) and cellular component (CC) of genes involved in the PPI network. Significant functions were plotted against –log10 of the p-value for up and down-regulated genes separately. In this analysis p-value <0.05 was set as the criterion.
Pathway enrichment analysis
Pathway enrichment of DEGs involved in the PPI network was also performed with DAVID. Significantly enriched KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways were selected and plotted against –log10 of the p-value. The criterion for this analysis was set as a p-value <0.05.
Known disease genes
Known and validated genes of GDM were downloaded from OMIM [18] and Gene Cards [19] and compared with DEGs to find candidate GDM-related genes. OMIM and Gene Cards provide information of all annotated and experimentally validated genes related to human disease. Co-enrichment analysis for these candidates GDM related gene and known disease gene was performed.
DEGs between specimens of GDM patients and healthy controls
Normalized microarray data of GDM and control specimens have been shown in Figure 1. A total of 571 significant DEGs with their official gene symbol were identified of which 302 were up-regulated and 269 were down-regulated. Criteria used for selection was p-value <0.05 and | log2 FC >0.1. DEGs were selected based on their average gene expression value. Moreover, 7 DEGs (3 up and 4 down) were found to be common in OMIM and Gene Cards.
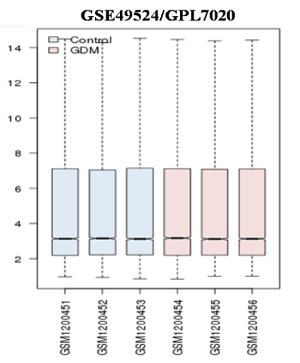
Figure 1. Microarray data after normalization. Box plot showing the distribution of values data for the selected samples. The lines in the box are coincident, indicating that these chips have been highly normalized
Principal component and hierarchical clustering analysis of DEGs
Principal Component Analysis reveals a scatter plot showing total variance of 75.9% and 7.2% with axes corresponding to the two different principal components - principal component 1 (x-axis) and principal component 2 (y-axis) respectively (Figure 2A). Heat-map shows a data matrix where coloring gives an overview of the numeric differences. The heat map was constructed for all 571 differentially expressed genes (Figure 2B).
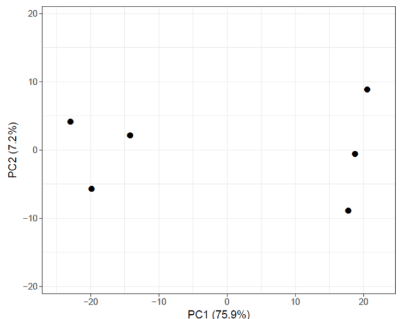
Figure 2A. Principal component analysis of dataset GSE49524. PCA plot shows a scatter plot with principal component 1 (x-axis) and principal component 2 (y-axis) showing total variance of 75.9% and 7.2% respectively. ClustVis tool was used for this
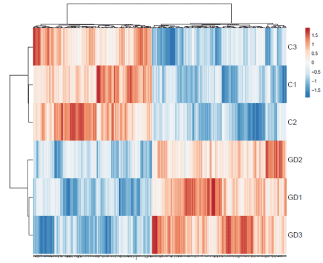
Figure 2B. Heat map of differentially expresses gene sets. Heat map showing the average gene expression of differentially expressed genes (DEGs) among gestational diabetes mellitus (GDM) and healthy control (HC). The blue to orange gradation represents the gene expression values change from small to large. ClustVis tool was used to draw heat map
The Protein-Protein interaction network
All DEGs with combined score >0.9 (170 gene pairs out of 571 DEGs) was used to form protein-protein interaction network which yielded one main network having 107 nodes and 179 edges (Figure 3) while five sub-networks were extracted separately (Figure 4). Out of a total of 170 DEGs with a combined score >0.9, a total of 96 DEGs were up-regulated (red node) while 74 were down-regulated (blue node) (Figure 5).
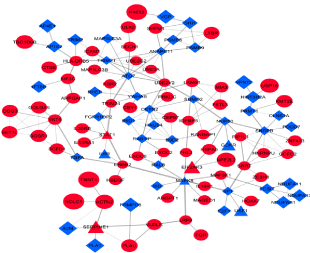
Figure 3. Protein-Protein interaction (PPI) of differentially expressed genes. Red Circle up-regulated genes, Blue rectangle down-regulated genes, Red and Blue triangle known disease genes. Lines the correlation between genes Thickness of lines (edges) is proportional to the combined score. Cytoscape v 3.2.1 was used to construct the network
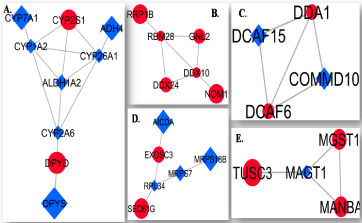
Figure 4. Five subnetworks of differentially expressed genes (DEGs). A. subnetwork 1. B. subnetwork 2. C. subnetwork 3. D. subnetwork 4, and E. subnetwork 5. Red Circle up-regulated genes, Blue rectangle down-regulated genes. Lines the correlation between genes (proteins). The thickness of the lines (edges) is proportional to the combined score. Cytoscape v 3.2.1 was used to extract subnetwork
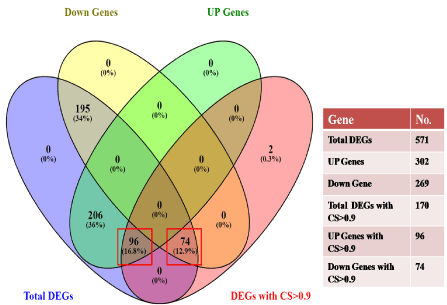
Figure 5. Venn diagram showing the total number of DEGs. DEGs with the combined score (CS) >0.9, total up-regulated genes, and down-regulated genes. The red rectangle highlights the total UP and DOWN regulated genes having a combined score >0.9. Venny tool v 2.1.0 was used to draw the vein diagram
Sub-network construction
Five sub-networks were extracted from the main network by the use of Cytoscape (Figure 4A-4E). Sub-network 1 has 9 nodes and 13 edges (Figure 4A), sub-network 2 has 6 nodes and 7 edges (Figure 4B), sub-network 3 has 6 nodes and 5 edges (Figure 4C), sub-network 4 has 4 nodes and 5 edges (Figure 4D), and sub-network 5 has 4 nodes and 4 edges (Figure 4E).
Known disease genes and candidate GDM related genes
Comparison of these 170 DEGs with GDM related annotated gene list obtained from OMIM and Gene Cards (Figure 6A) reveals 7 known disease genes (represented as a red and blue triangle in the main network, Figure 3). Direct neighbors of these known genes were selected as candidate GDM related genes; this yields a total of 8 candidate genes which are shown in Figure 6B. Although, none of the genes were significant except PLAT when plotted against average gene expression value (Figure 6D), however, at fold change level all these eight genes were found to be significant (p-value <0.05; Figure 6C). Out of these 8 candidate GDM genes, 3 up-regulated genes were IL13RA1 (Interleukin 13 Receptor Subunit Alpha 1), HSPA5 (Heat Shock Protein Family A (Hsp70) Member 5) and PLAU (Plasminogen Activator, Urokinase) while five down-regulated genes were TGFA (Transforming growth factor-alpha), PLAT (Plasminogen activator), BID (BH3 Interacting Domain Death Agonist) and SP1 (Upstream Transcription Factor 1) and NUP50 (Nucleoporin 50).
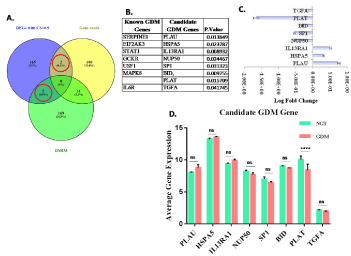
Figure 6. Known validated Genes: A. Vein diagram showing common genes among DEGs, OMIM, and Gene cards. B. List of known and candidate GDM related genes. C. Log Fold Change bar graph for candidate GDM gene. D. Bar graph showing average gene expression of candidate GDM gene
Functional enrichment analysis
Gene ontology enrichment analysis for DEGs involved in the PPI network was performed and significantly enriched functions, processes, and cellular components (p-value <0.05) were listed in Table 1 (For up-regulated DEGs) and Table 2 (For down-regulated DEGs). Among these biological processes, regulation of RAS protein signal transduction, regulation of apoptosis, cell proliferation, regulation of the immune system, regulation of secretion and wound healing were major significant processes (p-value <0.05) related to up-regulated DEGs (Figure 7A) while regulation of hormone levels, pro-apoptotic gene products, embryo and limb morphogenesis, neuronal cell development, pancreas development, oxidative phosphorylation and response to hypoxia were significant processes (p-value <0.05) of down-regulated genes (Figure 7B).
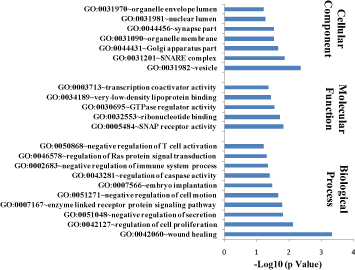
Figure 7A. Gene Ontology analysis for UP-regulated Genes in PPI network in diabetic mothers. Bar graph showing significant processes, function and cellular component enriched in diabetic mothers for up-regulated genes. DAVID v 6.7 was used for annotation

Figure 7B. Gene Ontology analysis for Down-regulated Genes in PPI network in diabetic mothers. Bar graph showing significant processes, function and cellular component enriched in diabetic mothers for down-regulated target genes. DAVID v 6.7 was used for annotation
Table 1. Gene Ontology and Pathway Enrichment Analysis of Up - regulated Genes
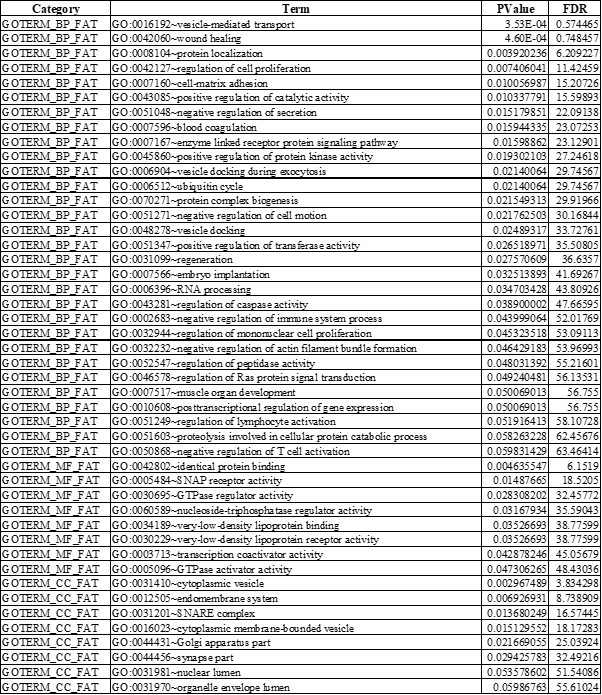
BP: Biological process; MF: Molecular function; CC: Cellular component; FDR: False Discovery Rate
Table 2. Gene Ontology and Pathway Enrichment Analysis of Down - regulated Genes
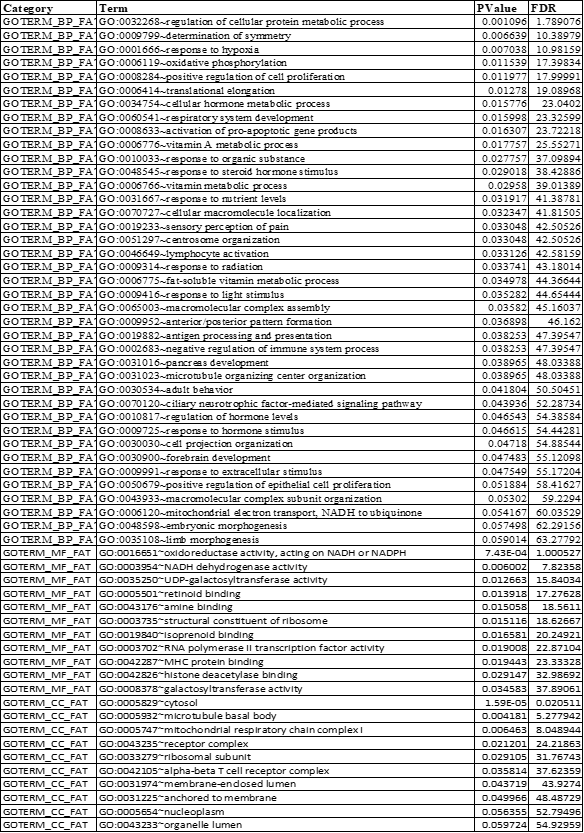
BP: Biological process; MF: Molecular function; CC: Cellular component; FDR: False Discovery Rate
Co-enrichment analysis for these candidates GDM related gene led to the identification of important processes which are listed in Table 3. A separate biological processes network was created for those eight candidates GDM related genes (Figure 8A and 8B). UP-regulated genes (IL13RA1, HSPA5, and PLAU) were found to be involved in the regulation of cell proliferation, caspase activity, wound healing, and enzyme-linked receptor signaling (Figure 8A). Major processes regulated by down-regulated genes (TGFA, PLAT, BID, SP1, and NUP50) were pancreas development, activation of pro-apoptotic genes, response to hypoxia, and protein localization (Figure 8B). Hence, 8 DEGs identified in this study were estimated to be candidate disease genes of GDM.
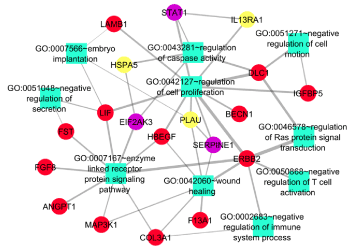
Figure 8A. Functional analysis of significantly Up-regulated genes. Functional analysis uncover many significant processes being regulated by the candidate and known GDM related genes. Red ellipse up-regulated genes, purple ellipse known GDM genes, Yellow ellipse candidate GDM related genes, Light green rectangle biological processes
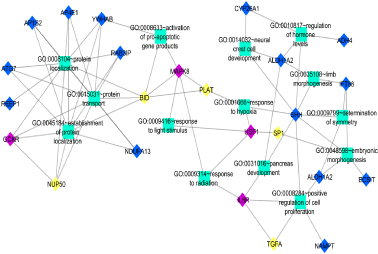
Figure 8B. Functional analysis of significantly Down-regulated genes. Functional analysis uncover many significant processes being regulated by the candidate and known GDM related genes. Red ellipse up-regulated genes, purple ellipse known GDM genes, Yellow ellipse candidate GDM related genes, Light green rectangle biological processes
Table 3. The comprehensive information of Candidate GDM DEGs enriched in significant functions
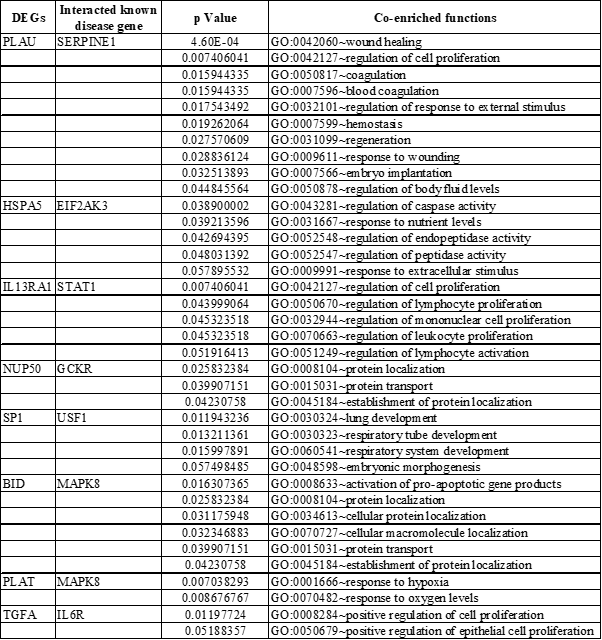
DEGs: Differentially Expressed Genes
Pathway enrichment analysis
KEGG pathway enrichment analysis for DEGs in the PPI network revealed a total of 6 significantly enriched pathways (p value<0.05). Among these pathways, oxidative phosphorylation, ribosome, pancreatic cancer, and glycosphingolipid biosynthesis are associated significantly with GDM (Figure 9A). Genes playing an important role in these pathways are listed in Figure 9B. It should be noted that IL13RA1, HSPA5, PLAU, TGFA, PLAT, BID, SP1, and NUP50 were enriched in GO functions and/or KEGG pathways together with their interacted known disease genes.

Figure 9A. KEGG Pathway analysis for Differentially Expressed Genes in diabetic mothers. Pathway enrichment for DEGs lead to identification of 2 significant pathways for up- while 4 significant pathways for down-regulated DEGs. DAVID v 6.7 was used for annotation. Red and blue bar represents pathways of up-and down-regulated genes respectively
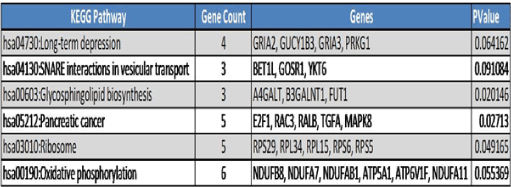
Figure 9B. Significantly enriched KEGG Pathways and genes involved in these pathways
In the past few years, GDM has become the global maternal health problem with the adverse fetal outcome and compromised maternal and neonatal health. Despite its increasing incidence, the main candidate gene and the complete molecular mechanism is not yet deciphered. In this microarray study, we have tried to find out the new candidate genes and pathways being involved in the prognosis of GDM. In this study, microarray data submitted on GEO datasets (GSE49524; Figure 1) was statistically analyzed to screen out DEGs. A total of 571 DEGs were identified of which 302 up-regulated genes and 269 down-regulated genes were identified (Figure 5). PPI networking for a total of 172 genes with a combined score>0.9 was performed (Figure 3). Functional enrichment analysis for genes in PPI network reveals regulation of cell proliferation, regulation of the immune system, regulation of caspase activity, RAS signaling, and regulation of secretion as major significant processes being regulated by the up-regulated genes (Figure 7A) while the response to hypoxia, oxidative phosphorylation, regulation of hormone levels, pancreas development, embryo morphogenesis, and neuronal cell development are significant major processes being regulated by the down-regulated genes (Figure 7B). Pathway enrichment analysis for these genes led to the identification of oxidative phosphorylation, ribosome, pancreatic cancer, and glycosphingolipid biosynthesis as major significant pathways (Figure 9A). Further, the PPIs network has been investigated between DEGs and known disease gene and functional consistency was assessed via functional enrichment analysis. Consequently, 8 candidate GDM-related genes IL13RA1, HSPA5, PLAU, TGFA, PLAT, BID, SP1, and NUP50 were identified.
Among these genes, IL13RA1 interacted with the STAT1-known GDM gene (Figure 6B and 8A) and they were enriched in regulation of cell proliferation. Higher expression of STAT1 has been reported in GDM cases and is essential for cytokine signaling [20]. IL13RA1 (IL-13Rα1) is a cytokine receptor and has been shown to play a role in macrophage differentiation and function [21] and its elevated expression cause neuronal loss during severe chronic stress [22]. In this study also, higher expression of IL13RA1 has been found (Figure 6C), confirming the chronic stress-induced diabetic milieu and increased oxidative stress. Furthermore, increased expression of IL-13Rα1 may also compromise fetal development by negatively regulating neuronal cell and macrophage proliferation.
HSPA5 interacted with known disease genes EIF2AK3 (Figure 6B and 8A), and they were enriched in the regulation of caspase activity and enzyme-linked receptor signaling pathway. An earlier study in mouse has reported that loss of function mutation in perk (EIF2AK3) causes insufficient proliferation of β-cells and defects in insulin secretion leading to neonatal diabetes in human (Wolcott Rallison Syndrome) and mice attributable to the impaired ER-to-Golgi anterograde trafficking, retrotranslocation from the ER to the cytoplasm, and proteasomal degradation. Hence, increased expression of EIF2AK3 hastens the progression of diabetes [23]. Another study reports the perk (EIF2AK3) as an essential regulator of the endoplasmic reticulum (ER) stress response [24] and ER stress has been found to promote β-cell apoptosis in type 2 diabetes patient [25]. Similarly, in this study, the expression of both of these genes are up-regulated, hence may be beneficial. No study reports the similar role of HSPA5 in any diabetes including GDM. Since HSPA5 directly interacts with known disease gene EIF2AK3 hence it will be playing a similar role and can be considered as a strong and novel candidate gene of GDM. However, further validations are required need to support these findings.
PLAU interacts with known disease gene SERPINE1, and they were found to be involved in wound healing and embryo implantation. In a study conducted by Sun H, et al., it has been shown that wounding up-regulates the expression of SERPINE1 and PLAU, and in healing epithelia, Plau and Serpine1 were abundantly expressed at the leading edge of the healing epithelia [26]. Another study reports increased expression of SERPINE1 in endometriosis [27] and lung cancer. In addition to its role in wound healing, SERPINE1 is also thought to be involved in cell movement (migration) and the breakdown and replacement (remodeling) of body tissues [28]. PLAU is a plasminogen activator while SERPINE1 is a plasminogen activator inhibitor, hence the action of both genes, balance the cell growth and movement during embryo development, and hence their expression may be detrimental to fetal health in hyperglycemic condition.
TGFA interacts with known disease gene IL6R and both are found to regulate cell proliferation and pancreas development. Various studies report the role of IL6/IL6R in pancreatic development and function. In one study, conducted by Wu X, et al. IL6R has been found to inhibit pancreatic β-cell apoptosis in type 2 diabetes via JAK-STAT signaling [29]. IL6/IL6R has also been found to regulate pancreatic cell mass expansion, particularly α-cell [30] and secretion of insulin by directly acting on pancreatic β-cell [31]. Further, TGFA has also been found to stimulate the growth of islet cells of pancreas [32,33]. However, both TGFA and IL6R are down-regulated in GDM cases and hence can be thought of as role players in the development of GDM by hampering the growth and function of the pancreas.
PLAT and BID both interact with known disease gene MAPK8 and they were found to respond to hypoxia and were found to be involved in activation of the pro-apoptotic gene product. MAPK8 is encoded mitogen-activated protein kinase important for apoptosis, T-cell differentiation, and inflammatory responses [34]. Increased expression of Jnk1 (Mapk8) is required for EMT cell migration and the induction of apoptosis [35]. BID has also been found in apoptosis of β-cell and its deficiency protects beta cells from FasL-induced apoptosis in vitro [36]. No study reports the role of PLAT in any kind of diabetes including GDM. Since PLAT interacts with BID and MAPK8, both of which play an important role in apoptosis of beta-cell, hence it can also be considered to play a similar role and thus may act as a novel candidate gene for GDM. However, further experimentation will be required to validate the findings.
SP1 interacts with known disease gene USF1 and both control the embryonic morphogenesis. Upstream stimulating factor 1 (USF1) is a basic helix loop helix transcription factor that regulates oocyte and early embryo development through its specific binding to E-box DNA motifs which are known cis-elements of key oocyte expressed genes [37]. Despite their role in embryo development, it has also been reported to play an important role in insulin synthesis. Furthermore, USF1 is found to be involved in the transcriptional regulation of various genes whose gene products are implicated in the stress and immune response, cell cycle regulation, DNA repair and proliferation of cells, and in lipid and carbohydrate metabolism [38]. Like USF1, Specificity protein 1 (Sp1) is also a transcription factor and has been found to regulate gene expression in response to insulin [39] not only this insulin has been found to stimulates both the biosynthesis of transcription factor Sp1 as well as its O-linked N-acetyl-glucosaminylation (O-GlcNAcylation) [40]. It also plays an important role in the developmental process of various organs like egg-laying system [41] including placenta [42]. Hence both genes play a diverse role and are particularly involved in embryo morphogenesis and insulin metabolism in case of diabetes.
NUP50 interacts with known disease gene GCKR and they were enriched in protein localization and protein transport. The glucokinase regulatory protein (GCKR) regulates the activity of the glucokinase (GCK) and has been shown to post-transcriptionally regulates GCK function in the liver, and causes its nuclear localization, not only this, but GCK also causes nuclear localization of GCKR [43]. Various association studies reveal the important role of GCKR in diabetes development [44,45]. NUP50 is a type of nuclear pore complexes (NPCs) that have long been known in the transport of protein and RNA between the nucleus and the cytoplasm [46]. Several studies report the universal role of NUP50 in nuclear protein export [47,48]. Hence both the genes are important for protein localization particularly glucokinase protein, in case of GDM, which is important in glucose metabolism.
The present study identifies 8 candidates GDM related genes based on the PPI network and functional consistency. Hence these genes namely IL13RA1, HSPA5, PLAU, TGFA, PLAT, BID, SP1, and NUP50 can be considered as a candidate gene for GDM based on their direct correlation with known disease gene and bio-functional consistency. Out of these 8 candidate genes, two genes namely PLAT and HSPA5 has not been reported in any diabetic condition including GDM so far to best of our knowledge and thus can be considered as a novel candidate gene of GDM. However, further experimentation will be required for the validation of these genes. Our study will contribute to the understanding of mechanisms lying behind the progression of GDM. Nevertheless, more studies are required to validate our predictions.
Ravi Bhushan: Methodology, investigation, writing - review & editing.
Anjali Rani: Visualization, investigation, validation.
Deepali Gupta: Visualization, validation.
Vinay Kumar Singh: Data curation, software.
Pawan K. Dubey: Conceptualization, writing - review & editing.
The authors are highly thankful to all study participants and acknowledge their valuable help for this study.
The authors declare that they have no conflict of interest.
- Bhushan R, Dubey PK (2018) Gestational diabetes mellitus: Risk and its challenges. J Mol Genet Med 12: 1000340.
- Poulakos P, Mintziori G, Tsirou E, Taousani E, Savvaki D, et al. (2015) Comments on gestational diabetes mellitus: from pathophysiology to clinical practice. Hormones 14: 335-344. [Crossref]
- Bevier WC, Jovanovic-Peterson L, Peterson CM (1995) Pancreatic disorders of pregnancy: Diagnosis, management, and outcome of gestational diabetes. Endocrinol Metab Clin North Am 24: 103-138. [Crossref]
- American Diabetes Association (2010) Diagnosis and classification of diabetes mellitus. Diabetes Care 33: S62-S69. [Crossref]
- Agarwal MM, Dhatt GS, Shah SM. Gestational Diabetes Mellitus. Diabetes Care 33: 2018-2020. [Crossref]
- Buchanan TA, Xiang AH (2005) Gestational diabetes mellitus. J Clin Invest 115: 485-491. [Crossref]
- Ashwal E, Hod M (2015) Gestational diabetes mellitus: Where are we now? Clinica Chimica Acta 451: 14-20. [Crossref]
- Wendland EM, Torloni MR, Falavigna M, Trujillo J, Dode MA, et al. (2012) Gestational diabetes and pregnancy outcomes - a systematic review of the World Health Organization (WHO) and the International Association of Diabetes in Pregnancy Study Groups (IADPSG) diagnostic criteria. BMC Pregnancy Childbirth 12: 23. [Crossref]
- Cooper ME, El-Osta A (2010) Epigenetics: Mechanisms and implications for diabetic complications. Circ Res 107: 1403-1413. [Crossref]
- Ambra R, Manca S, Palumbo MC, Leoni G, Natarelli L, et al. (2014) Transcriptome analysis of human primary endothelial cells (HUVEC) from umbilical cords of gestational diabetic mothers reveals candidate sites for an epigenetic modulation of specific gene expression. Genomics 103: 337-348. [Crossref]
- Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, et al. (2007) NCBI GEO: mining tens of millions of expression profiles--database and tools update. Nucleic Acids Res 35: D760-D765. [Crossref]
- Bhushan R, Rani A, Ali A, Singh VK, Dubey PK (2020) Bioinformatics enrichment analysis of genes and pathways related to maternal type 1 diabetes associated with adverse fetal outcomes. J Diabetes Complications 34: 107556. [Crossref]
- Davis S, Meltzer PS (2007) GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23: 1846-1847. [Crossref]
- Metsalu T, Vilo J (2015) ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res 43: W566-W570. [Crossref]
- Franceschini A, Szklarczyk D, Frankild S, Michael Kuhn, Milan Simonovic, et al. (2012) STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41: D808-D815. [Crossref]
- Kohl M, Wiese S, Warscheid B (2011) Cytoscape: Software for visualization and analysis of biological networks. Methods Mol Biol 696: 291-303. [Crossref]
- Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44-57. [Crossref]
- Amberger JS, Hamosh A (2017) Searching online mendelian inheritance in man (OMIM): A knowledgebase of human genes and genetic phenotypes. Curr Protoc Bioinformatics 58: 1-12. [Crossref]
- Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, et al. (2016) The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics 54: 1-33. [Crossref]
- Kuzmicki M, Telejko B, Wawrusiewicz-Kurylonek N, Lipińska D, Pliszka J, et al. (2013) The expression of genes involved in NF-κB activation in peripheral blood mononuclear cells of patients with gestational diabetes. Eur J Endocrinol 168: 419-427.
- Dhakal M, Hardaway JC, Guloglu FB, Miller MM, Hoeman CM, et al. (2014) IL-13Rα1 is a surface marker for M2 macrophages influencing their differentiation and function: Innate immunity. Eur J Immunol 44: 842-855.
- Mori S, Sugama S, Nguyen W, Michel T, Sanna MG, et al. (2017) Lack of interleukin-13 receptor α1 delays the loss of dopaminergic neurons during chronic stress. J Neuroinflammation 14: 88. [Crossref]
- Gupta S, McGrath B, Cavener DR (2010) PERK (EIF2AK3) Regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes 59: 1937-1947. [Crossref]
- Shi W, Xu G, Wang C, Sperber SM, Chen Y, et al. (2015) Heat shock 70-kDa protein 5 (Hspa5) is essential for pronephros formation by mediating retinoic acid signaling. J Biol Chem 290: 577-589. [Crossref]
- Laybutt DR, Preston AM, Åkerfeldt MC, Kench JG, Busch AK, et al. (2007) Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 50: 752-763. [Crossref]
- Sun H, Mi X, Gao N, Yan C, Yu F-S (2015) Hyperglycemia-suppressed expression of Serpine1 contributes to delayed epithelial wound healing in diabetic mouse corneas. Invest Ophthalmol Vis Sci 56: 3383. [Crossref]
- Alotaibi FT, Peng B, Klausen C, Lee AF, Abdelkareem AO, et al. (2019) Plasminogen activator inhibitor-1 (PAI-1) expression in endometriosis. PLoS ONE 14: e0219064. [Crossref]
- Yildiz YS, Kuru P, Oner ET, Agirbasli M (2014) Functional stability of plasminogen activator inhibitor-1. Scientific World Journal 2014: 1-11. [Crossref]
- Wu X, Yu T, Ji N, Huang Y, Gao L, et al. (2019) IL6R inhibits viability and apoptosis of pancreatic beta-cells in type 2 diabetes mellitus via regulation by miR-22 of the JAK/STAT signaling pathway. Diabetes Metab Syndr Obes 12: 1645-1657. [Crossref]
- Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, et al. (2008) Interleukin-6 regulates pancreatic α-cell mass expansion. Proc Natl Acad Sci U S A 105: 13163-13168. [Crossref]
- Suzuki T, Imai J, Yamada T, Ishigaki Y, Kaneko K, et al. (2011) Interleukin-6 enhances glucose-stimulated insulin secretion from pancreatic -cells: Potential involvement of the PLC-IP3-dependent pathway. Diabetes 60: 537-547.
- Wang TC, Bonner-Weir S, Oates PS, Chulak M, Simon B, et al. (1993) Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. J Clin Invest 92: 1349-1356. [Crossref]
- Wang RN, Rehfeld JF, Nielsen FC, Klöppel G (1997) Expression of gastrin and transforming growth factor-α during duct to islet cell differentiation in the pancreas of duct-ligated adult rats. Diabetologia 40: 887-893. [Crossref]
- Zhang X, Zhang W, Ma S-F, Desai AA, Saraf S, et al. (2014) Hypoxic response contributes to altered gene expression and precapillary pulmonary hypertension in patients with sickle cell disease. Circulation 129: 1650-1658.
- Tiwari N, Meyer-Schaller N, Arnold P, Antoniadis H, Pachkov M, et al. (2013) Klf4 Is a Transcriptional Regulator of Genes Critical for EMT, Including Jnk1 (Mapk8). PLoS ONE 8: e57329. [Crossref]
- Spohrer S, Groß R, Nalbach L, Schwind L, Stumpf H, et al. (2017) Functional interplay between the transcription factors USF1 and PDX-1 and protein kinase CK2 in pancreatic β-cells. Sci Rep 7: 16367.
- Datta TK, Rajput SK, Wee G, Lee K, Folger JK, et al. (2015) Requirement of the transcription factor USF1 in bovine oocyte and early embryonic development. Reproduction 149: 203-212. [Crossref]
- Mollah ZUA, Wali J, McKenzie MD, Krishnamurthy B, Graham KL, et al. (2011) The pro-apoptotic BH3-only protein Bid is dispensable for development of insulitis and diabetes in the non-obese diabetic mouse. Apoptosis 16: 822-830.
- Solomon SS, Majumdar G, Martinez-Hernandez A, Raghow R (2008) A critical role of Sp1 transcription factor in regulating gene expression in response to insulin and other hormones. Life Sciences 83: 305-312. [Crossref]
- Majumdar G, Wright J, Markowitz P, Martinez-Hernandez A, Raghow R, et al. (2004) Insulin stimulates and diabetes inhibits O-Linked N-Acetylglucosamine Transferase and O-Glycosylation of Sp1. Diabetes 53: 3184-3192. [Crossref]
- Ulm EA, Sleiman SF, Chamberlin HM (2011) Developmental functions for the caenorhabditis elegans Sp protein SPTF-3. Mech Dev 128: 428-441. [Crossref]
- Li M, Kellems RE (2003) Sp1 and Sp3 are important regulators of AP-2 gene transcription. Biol Reprod 69: 1220-1230. [Crossref]
- Jin L, Guo T, Li Z, Lei Z, Li H, et al. (2015) Role of glucokinase in the subcellular localization of glucokinase regulatory protein. Int J Mol Sci 16: 7377-7393. [Crossref]
- Lin Z, Wang Y, Zhang B, Jin Z (2018) Association of type 2 diabetes susceptible genes GCKR, SLC30A8, and FTO polymorphisms with gestational diabetes mellitus risk: a meta-analysis. Endocrine 62: 34-45. [Crossref]
- Mohás M, Kisfali P, Járomi L, Maász A, Fehér E, et al. (2010) GCKR gene functional variants in type 2 diabetes and metabolic syndrome: do the rare variants associate with increased carotid intima-media thickness? Cardiovasc Diabetol 9: 79. [Crossref]
- Smitherman M, Lee K, Swanger J, Kapur R, Clurman BE (2000) Characterization and targeted disruption of murine Nup50, a p27Kip1-interacting component of the nuclear pore complex. Mol Cell Biol 20: 5631-5642. [Crossref]
- Makise M, Mackay DR, Elgort S, Shankaran SS, Adam SA, et al. (2012) The Nup153-Nup50 protein interface and its role in nuclear import. J Biol Chem 287: 38515-38522. [Crossref]
- Guan T, Kehlenbach RH, Schirmer EC, Kehlenbach A, Fan F, et al. (2000) Nup50, a nucleoplasmically oriented nucleoporin with a role in nuclear protein export. Mol Cell Biol 20: 5619-5630.