The study of coronary artery disease (CAD) has evolved throughout the years, specially aiming at the identification of risk factors. Traditional risk factors include high cholesterol levels , high blood pressure, diabetes mellitus, obesity, inflammation, age, and sex. Additionally, the study of monogenic models of CAD, such as familial hypercholesterolemia, represented a landmark in the understanding of the genetic contribution to CAD. The incorporation of novel techniques, such as GWAS (genome-wide association study), has allowed the identification of specific genetic features that contribute to CAD. GWAS is a method that involves scanning the genome from several individuals of a set population, to look for genetic markers that can be related to a higher risk of disease. Several SNP (single nucleotide polymorphisms), have been identified in the genes involved in the cholesterol metabolism pathway. The most studied locus linked to atherosclerosis is chromosome 9p21.3. Initially, the study of this risk variant was made with individual or familial studies, whereas to date, the creation of GWAS consortiums has allowed a more detailed study. In this review we discuss the role of chromosome 9p21.3 polymorphisms and the main risk variants identified. Despite the big amount of research in European, Asiatic and North American populations, there is a considerable gap of knowledge in the Latin American population, in which no SNP of relevance has been found. The study of risk variants in the future may allow earlier implementation of primary and secondary prevention strategies.
Coronary artery disease (CAD), is characterized by the chronic development of atherosclerotic plaques in the walls of coronary arteries. It is considered a chronic inflammatory process that evolves throughout the life of individuals since the moment of birth and is intimately associated with primary and secondary risk factors [1]. The formation and subsequent growth of these plaques continues at a constant rate resulting in the shrinkage of the arterial lumen. This may cause myocardial ischemic events, which pose a huge risk to the life of patients [2].
The latest estimates state that more than 126 million people around the world are affected by this disease. This represents 1.72% of the global population, which places the prevalence at 1,655 cases per 100,000 people. The male-female ratio exhibits an increased risk for male patients as they approach middle age. By the year 2030, an increase in prevalence, from 1,655 to 1,845 cases per 100,000, is expected. Geographically, the area with the highest prevalence lies in Eastern Europe [3].
The formation of atherosclerotic plaques within the arterial lumen results in deficient gas and nutrient exchange, which increases myocardial stress and reduces contractility. Structural changes in the tunica intima lead to alterations in the nitric oxide (NO) pathway and impair the vessel’s contractile ability [2]. The damage to the tunica intima enables the infiltration of the tunica media by low-density lipoproteins (LDL), which increases pro-inflammatory cytokine activity, particularly IL-1. This process stimulates the arrival of innate immune cells, particularly macrophages. Such cells phagocyte LDL molecules, which results in apoptosis and, finally, epherocytosis. The buildup of LDL, innate immune cells, and macrophages constitutes the atherosclerotic plaque. This process becomes a vicious cycle that ends in ischemic or hemorrhagic events [3].
Thanks to genetic studies of CAD, the 9p21 locus has been associated with an approximately 30% increased risk of CAD per copy of the risk allele [2]. Furthermore, with the help of Genome Wide Association Studies (GWAS), larger population samples have been studied to look for the genetic backbone of CAD, yielding approximately 60 distinct genetic loci. Nearly 20% of the loci are located close to genes with significant roles in the metabolism of LDLs, triglycerides, and lipoprotein(a) (a modified LDL particle) [3].
Family-based studies have allowed the identification of genetic variants associated with CAD. [2] Familial hypercholesterolemia, which encompasses increased cholesterol and higher risk of CAD, was first related to a deletion in the gene encoding the LDL receptor, representing the first demonstration of a molecular defect in a single gene that leads to CAD [3]. Afterwards mutations in other genes were also identified, such as APOB (encodes apolipoprotein B), PCSK9 (encodes proprotein convertase subtilisin/kexin type 9), LDLRAP1 (encodes low-density lipoprotein receptor adapter protein 1) and ABCG5, ABCG8 (ATP-binding cassette sub-family G members 5 or 8) [4].
High blood pressure (BP) is a crucial risk factor for CAD, even after the implementation of antihypertensive therapy [4]. Regardless of the pharmacological treatment, a BP goal lower than 140/90 mmHg, is associated with reduced morbidity and mortality [5]. The SPRINT study and subsequent analysis led to the conclusion that systolic and diastolic pressure variability increases the risk of cardiovascular events (fatal and non-fatal) [6,7]. The United Kingdom Prospective Diabetes Study (UKPDS) showed that hypertension control in patients with type 2 Diabetes Mellitus (DM-2) reduces the risk of cardiovascular events (P <0.001), independently of the HbA1c level [8]. Furthermore, in the meta-analysis by Gorst C, et al. higher HbA1c variability was associated with higher risk of kidney disease (OR 1.34 [1.15-1.57], macrovascular events (OR 1.21 [1.06-1.38]), ulceration/gangrene (OR 1.50 [1.06-2.12]), cardiovascular disease (OR 1.27 [1.15-1.40]), and mortality (OR 1.34 [1.18-1.53]) [9].
The level of basal blood glucose and its variability are independent factors for the formation of atheromatous plaques, mainly due to endothelial stress [10]. The ADVANCE study analysis showed that glycemic variability is not associated with microvascular events in DM-2, vascular events (P = 0.01), or mortality (P <0.001). Variability produces greater stress and confers higher cardiovascular risk [10,11].
Based on epidemiology, an estimated 11-16% increase in cardiovascular events is observed for every 1% increase in HbA1c [10]. In patients with hypertension and diabetes, the latter is the most important factor to control for risk reduction [10-12].
Regarding smoking, the cardiovascular risk is proportional to the number of cigarettes smoked per day, increasing 1.48 for one (95% CI 1.30 to 1.69), 1.58 (1.39 to 1.80) for five, and 2.04 (1.86 to 2.24) for 20 cigarettes [13,14]. Taking a traditional mean of 3 cigarettes for men and 6 for women represents a 10-15% increased risk for CAD. Fortunately, quitting smoking reduces the risk of myocardial infarction by 50%. [13].
Family history is also an independent risk factor for CAD, with a predominant role in young patients [15,16]. In the ARIC study, familiar history resulted in a 17% increased risk of CAD ([HR]: 1.17; [95% CI]: 1.09 to 1.26; p <0.001) [16]. According to the NHANES survey, the risk increased by 12.2% in the case of a first-degree relative with cardiovascular disease (CVD) before the age of 50 [17].
Bodyweight variability also increases coronary events. Obesity acts as a risk factor for insulin resistance, diabetes, hypertension, dyslipidemia, heart failure, and CAD [18]. Obesity accelerates the progression of atherosclerosis and contributes to endothelial dysfunction and subclinical inflammation [19]. Reduction in insulin sensitivity, increased free fatty acid turnover, increased basal sympathetic tone, and a hypercoagulable state are observed in obesity [20]. By controlling cholesterol intake and smoking habits, as well as sudden significant weight loss, the risk decreases [21-23].
Finally, genetic variants have been recently described as a crucial risk factor for the development of CAD. Regarding the genetic study of CAD, the 9p21 locus is associated with an approximately 30% increased risk of CAD per copy of the risk allele [2]. Furthermore, with the help of GWAS studies, larger population samples have been studied to investigate the genetic backbone of CAD, yielding approximately 60 distinct genetic loci. Nearly 20% of the loci are located close to genes with significant roles in the metabolism of LDLs, triglycerides, and lipoprotein(a) (a modified LDL particle) [3]. To name a few, variants at the GUCY1A3 and NOS3 loci have been associated with both blood pressure and CAD [4].
Multiple rare genetic variants with a great influence on the pathophysiology of CAD have been elucidated. These variants could make us consider different subtypes of CAD. The combination of common genetic variants and environmental factors increases the risk of atherosclerotic plaque formation [1] (Table 1).
Table 1. Risk factors in coronary artery disease
Risk factors |
Generalities |
High blood pressure |
Systolic and diastolic pressure variability increases the risk of CAD. |
Type 2 diabetes mellitus |
Higher HbA1c variability was associated with cardiovascular disease (OR 1.27 [1.15-1.40]). |
Smoking |
Taking a traditional mean of 3 cigarettes for men and 6 for women represents a 10-15% risk for CAD. |
Family history |
The risk increased by 12.2% in the case of a first-degree relative with cardiovascular disease (CVD) before the age of 50. |
Body weight variability |
Obesity accelerates the progression of atherosclerosis and contributes to endothelial dysfunction and subclinical inflammation. |
Genetic predisposition |
9p21 locus polymorphisms, inactivating mutations conferring an increased risk (LDLR, LPL, APOA5) and a decreased risk (PCSK9, NPC1L1, ASGR1, APOC3, ANGPTL4 and lipoprotein (a)) |
Due to the high prevalence of cardiovascular diseases, several cardiovascular risk calculators have been developed. The Framingham calculator provides a general estimate, whereas the ATP III hard CHD risk score (2001) has a 10 year lifetime, and ultimately, the Spanish adaptation, which has validity during the whole patient's lifetime. Moreover, it can be estimated in low incidence countries [26-28]. However, these calculators do not take into account the genetic risk factors that have recently been elucidated. The principal characteristics of the most known cardiovascular risk calculators are summarized in Table 2.
Table 2. Examples of different cardiovascular risk calculators
CV risk calculator |
Variables |
Framingham [24]. |
Smoking, hypertension, high serum cholesterol, and familial history, integrated with age, sex, total cholesterol, HDL and presence of diabetes. |
Atp III hard CHD risk score [23]. |
Gender, age, smoking, systolic BP and total cholesterol or its ratio with HDL. Risk divided in 3 groups:
- 20% risk of heart disease
- Patients with diabetes.
- Patients with metabolic syndrome. |
ACC/AHA pooled cohort hard CVD risk Calculator [25] |
Age (40 to 75 years), presence of diabetes, sex, race (African American and white), total cholesterol, SAP and treatment for hypertension. |
The genetic field of CAD is complex and requires multiple approaches to visualize the factors that contribute to an individual's susceptibility to the development of specific diseases. Initially, thanks to genetic familiar studies, a small number of monogenic variables were found (for example, the genes initially associated with familial hypercholesterolemia) [42-46]. These variants were related to CAD with a premature onset. However, this mechanism was unable to explain all the genetic processes that generate CAD. Due to this gap of knowledge, it was necessary to make population studies that compared specific variations in the patients’ genome. In 2007, the first Genome-Wide Association Study (GWAS) was published [29].
Certain genome variants occur with a frequency of ≥0.5% in a population. They are known as polymorphisms and are relatively common [30]. Polymorphisms are classified into three groups. The first group involves changes in a single nucleotide variant (SNV), regarded as a change in nucleotide that occurs in less than 1% of the population, and single nucleotide polymorphism (SNP), which occurs in more than 1% of the population. The second group involves variations in the number of short DNA repetitive sequences. Finally, the third group includes variations in large DNA sequences (more than 1000 base pairs) [31].
The methodology of the GWAS studies is sequential. The first step consists in obtaining samples from individuals not related to one another, but that belong to the same population. Samples are analyzed using microarrays on chips that contain up to one million DNA markers. These markers consist of specific polymorphisms. Once these are identified, each variant is analyzed by comparing its frequency in the cases (people with the disease) against the controls (people without disease) [32]. GWAS allows the determination of which polymorphisms are more frequent with statistical significance in any given disease.
The results of GWAS studies are represented on a Manhattan plot. Unlike the traditional cut to demonstrate statistical significance (p <0.05), in the study of polymorphisms another value is needed: p <5 x 10-8. Because a large number of SNPs — whose individual effects are low — are included in GWAS, it must be adjusted for the number of polymorphisms tested using the Bonferroni correction, which is a method of controlling the overall probability of a false significant result when multiple comparisons are being performed. [33]. This type of study requires a very rigorous significance threshold because most DNA variants that have small effects require a lot of people to be studied to perform a successful GWAS [34]. The advantage of GWAS is that it surveys a broader gene pool compared with other related studies. In addition, it identifies the most relevant polymorphisms in the genome, to determine the existence of inheritance patterns [35].
Thanks to GWAS methodology (Figure 1) it has been determined that genetic predisposition is a combination of multiple variants, and each of these has a minimal effect on the phenotype [34]. Furthermore, it was determined that all of these variants represent 30-40% of heritability in CAD, with an increased risk of less than 20% [36]. Interestingly, the identified polymorphisms are outside of the protein-encoding regions [2], thus, they are regulatory variables that modify genes or pathways related to the development of this disease.
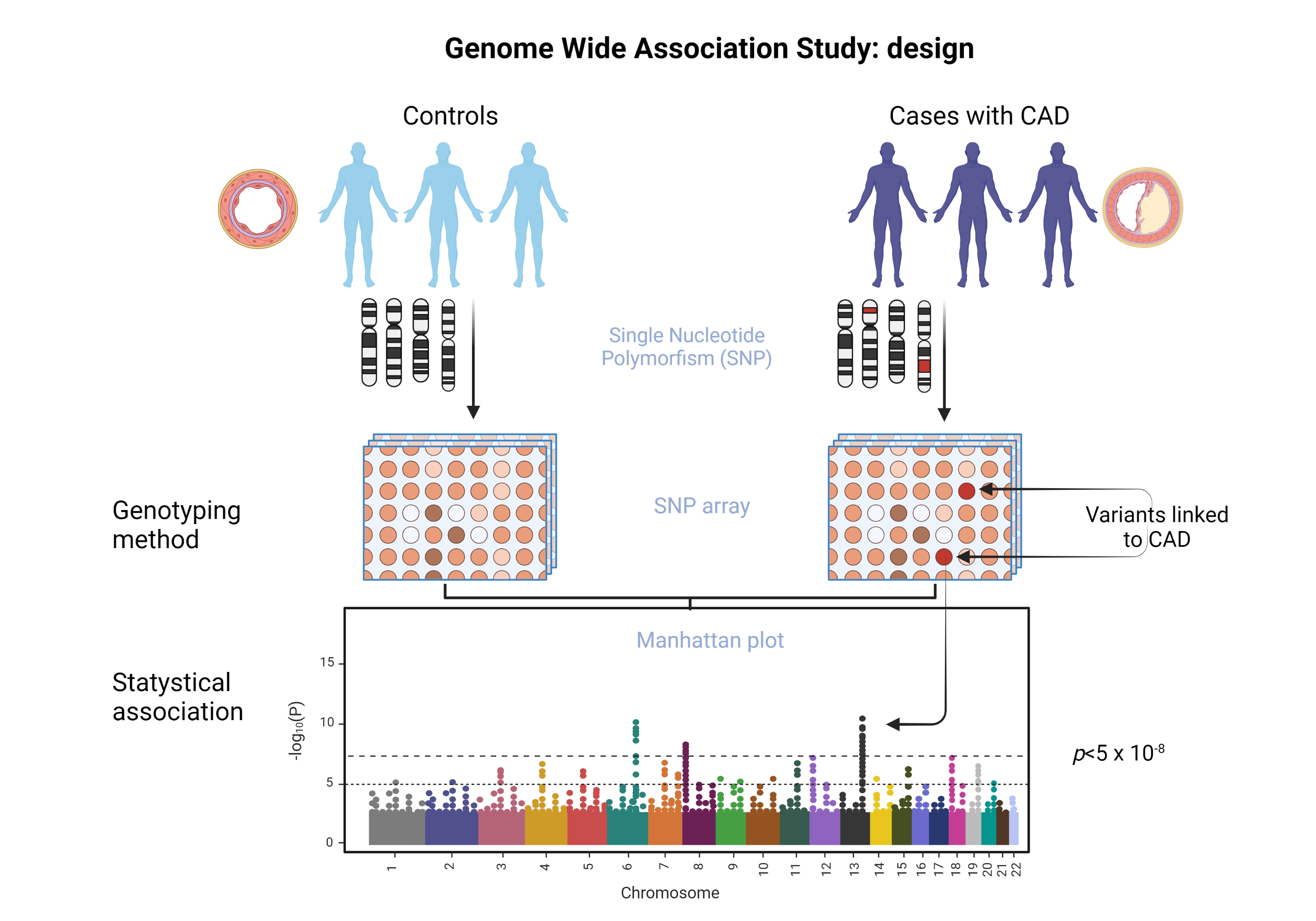
Figure 1. GWAS studies methodology
Monogenic inheritance
The variation in one gene that causes a phenotypic change and has a mendelian pattern corresponds to monogenic inheritance (Figure 2), which is considered the simplest form of inheritance. The study of this type of disease has been possible thanks to familial association studies [37]. Examples of this group are the monogenic risk variants for familial hypercholesterolemia, such as mutations in LDLR, APOB and PCSK9 and those related to a high content of triacylglycerols in lipoproteins, such as LPL and APOA5 [38,39].
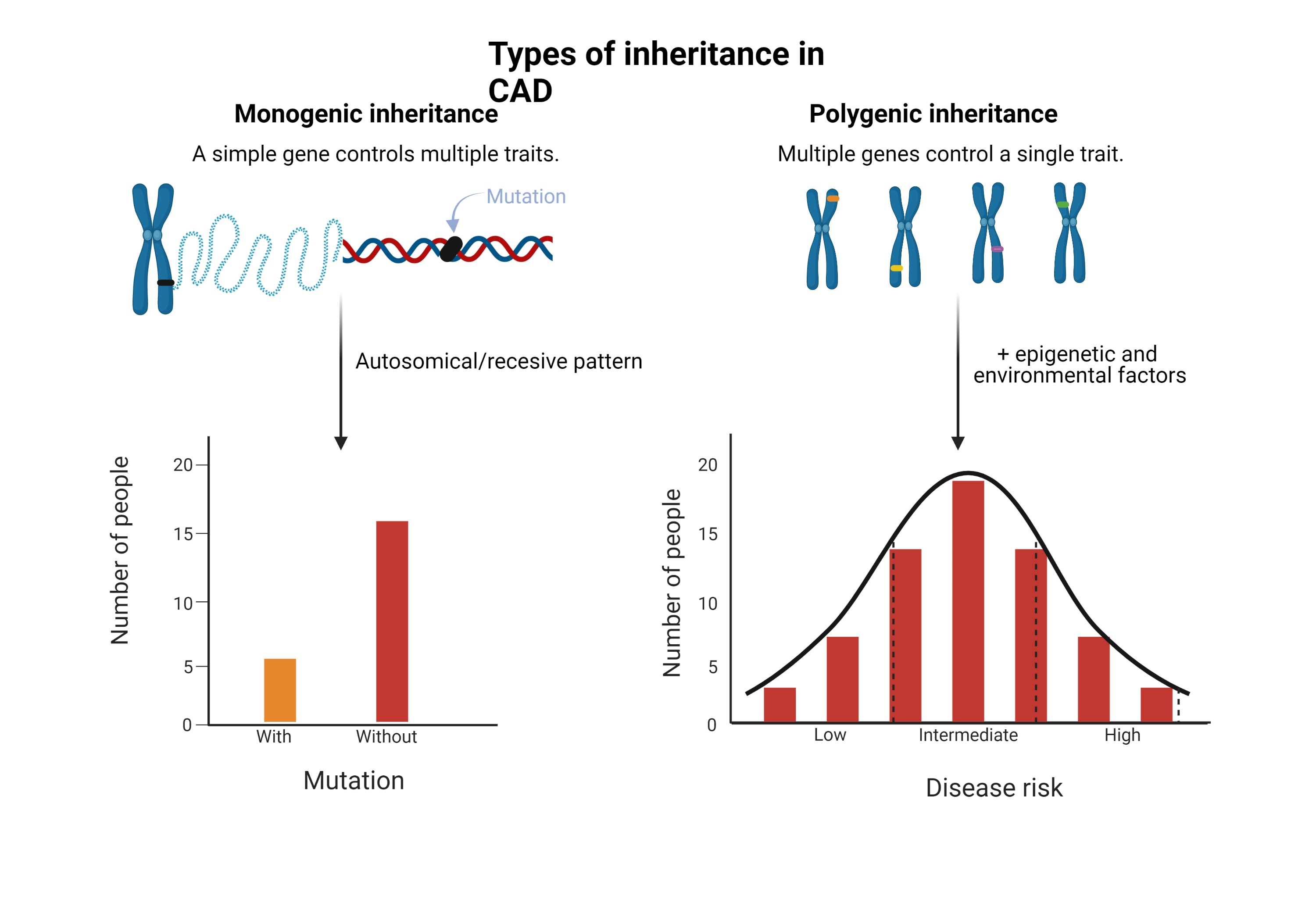
Figure 2. Types of inheritance in coronary artery disease
Polygenic inheritance
Most cardiovascular diseases depend on the interactions between multiple genetic variants and environmental factors. These are called “complex” or “multifactorial diseases” and have a polygenic inheritance (Figure 2) [40]. These types of diseases are the result of common variants with small individual effects combined with rare variants with moderate to strong effects [37]. Likewise, complex interactions are added to genetic, epigenetic, and environmental levels [41]. All genetic risk variants for CAD are common, 50% occur in more than half of the population and 25% occur in more than 75% of the population. Their relative risk is typically minimal to moderate, with an average increase of 18% in relative risk and with an odds ratio from 2% to 90% [34]. The fact that CAD has an inheritance with a strong environmental component, indicates that actions such as intensive lifestyle modification, early drug therapy, detection of subclinical disease, and genetic screening could improve the prognosis and quality of life.
There are more than 150 genes related to CAD. Although some of these genes are associated with abnormal lipid regulation, insulin resistance, clotting, inflammation, and vascular tone, most of the mechanisms of action are unknown [27]. Heritability is the product of many common genetic variants (frequency of minor alleles greater than 5%) that together with environmental factors have a significant role in the development of CAD. Thanks to GWAS, 20 common genetic variants have been hitherto identified [28].
Patients with familial heterozygous hypercholesterolemia without treatment have LDL-C levels from 150 to 500 mg/dl and more than 500 mg/dl in homozygous forms of the disease, with severe clinical presentation. About 5% of myocardial infarctions in patients under 60 years old and as many as 20% under the age of 45 are due to familial hypercholesterolemia (FH) [42]. The prevalence of its heterozygous form is 1: 500 and its homozygous form is 1: 1,000,000 [43]; a 6-fold increase in risk for CAD is observed in patients with LDL >190 mg/dL and no FH mutation (OR: 6.0; 95% CI: 5.2 to 6.9). Meanwhile, patients with LDL levels >190 mg/dl and an FH mutation have a 22-fold increased risk (OR: 22.3; 95% CI: 10.7 to 53.2) [2,44].
Three genetic variants are responsible for most cases. The first variant is the alteration of the LDL receptor leading to a lack of capacity to clear LDL from the circulation. This may be due to a lack of synthesis, incorrect transport from the endoplasmic reticulum to the Golgi apparatus, inadequate binding to LDL, incorrect endocytosis, or lack of recycling [42,45]. There can also be variants in the apolipoprotein B100 gene, which alters LDL receptor binding; variants in PCSK9, which normally lowers the lysosomal degradation of LDL-C; and variants in the gene that encodes apolipoprotein E, which directs the elimination of chylomicrons remnants and VLDL [45].
Alterations in LDLR genes represent 80-85% of familial hypercholesterolemia cases; apoB100 is responsible for 5-10% cases, PCSK9 for 2%, and LDL receptor adapter protein 1 (LDLRAP1) for less than 1%. Other uncommon involved genes are APOE, STAP1, LIPA, ABCG5, or ABCG8 [42,43].
Familial hypercholesterolemia can be diagnosed by clinical (Table 3) or molecular criteria. The latter facilitates the diagnosis but is not widely available due to their high cost [38] (Table 4).
Table 3. Diagnostic criteria of familial hypercholesterolemia
Study groups |
Criterion |
MEDPED USA |
Total LDL-C:
- <18 years: >270 mg/dl.
- 20-29 years: >290 mg/dl.
- 30-39 years: 340 mg/dl.
- >41 years: >360 mg/dl.
|
Dutch lipid clinic network |
Plasma LDL-C levels >4 mmol/L.
Family history of a first degree relative with premature CHD, LDL-C >95th percentile by age and gender for country (or a child <18 years), xanthoma and/or arcus cornealis.
Tendon xanthoma or arcus cornealis <45 years old.
Clinical history of premature ASCVD.
DNA analysis of mutation in LDLR, APOB, PCSK9. |
British Simon Broomea registry |
Total cholesterol >290 mg/dl (adult) or >260 mg/dl (child <16 years) or LDL-C >190 mg/dl (adult) or >155 mg/dl (child <16 years).
Tendon xanthomas in the patient or a first or second degree relative.
DNA-based evidence of genetic variants in LDLR, APO-B 100, PCSK9.
Family history of MI in a first-degree relative before the age of 60, or in a second-degree relative before age 50.
Family history in any first- or second-degree relative of a total plasma cholesterol level >290 mg/dl in an adult or >260 mg/dL in a child or sibling younger than 16 years old. |
MI (myocardial infarction), CHD (coronary heart disease), ASCVD (atherosclerotic cardiovascular disease), LDL-C (low density lipoprotein cholesterol), APOB (apolipoprotein B), PCSK9 (proprotein convertase subtilisin/kexin type 9).
Table 4. Risk variants of polymorphisms rsrs1333049 and rs10757278 and correlation with cardiovascular disease [57,58]
rs1333049 |
|
Genotype |
Population |
OR (95% CI) |
End study composite |
CC homozygous |
Russian |
OR 1.77 (95% CI 1.36–2.37) |
MI risk |
CG heterozygous |
Russian |
OR 1.58 (95% CI 1.18–2.11) |
MI risk |
GG homozygous |
Turkish |
OR 1.81 (95% CI 1.05-3.12) |
CAD risk |
rs10757278 |
GG homozygous |
Russian |
OR 1.70 (95% CI 1.24–2.32) |
MI risk |
CG heterozygous |
Russian |
OR 1.36 (95% CI 1.01–1.83) |
MI risk |
The introduction of GWAS studies in the field of research of CAD led to the identification of several loci in chromosome 9p21 in 2007. The main relevance of these loci is that they explain 30-40% of the inheritance capability of CAD, which has been estimated to cause 40% of cases [28,43,46,47].
Thanks to the formation of GWAS consortiums, like CARDIoGRAM, MIGen, C4D, and UK Biobank, a much larger number of patients have been studied, which has raised the number of identified loci [29]. Recent studies have analyzed the data provided by each consortium, leading to the typification of more than 60 risk loci associated with CAD.
More than 300 suggestive CAD loci have been found, which could be useful in the understanding of the biology and enhancement of the risk prediction of CAD [2,29]. Through the combination of 2 groundbreaking GWAS studies, CARDIoGRAM and C4D Consortium, 63 746 CAD cases and 130 681 controls were obtained, from which 153 loci linked to CAD were determined (46 significant loci with a false discovery rate of 5%). This represents a 10.6% inheritance of CAD [47]. Furthermore, these studies have allowed the creation of genetic risk scores, in order to calculate individual genomic risk. This type of genetic analysis aids in the calculation of CAD risk, with the advantage of being independent of other acquired risk factors [35].
Relevant identified pathophysiological pathways
The loci associated with CAD have been linked to pathological pathways related to atherosclerosis. The exact mechanisms through which the loci cause disease have not been completely identified.
The biology of the arterial wall has a crucial role in the pathogenesis of CAD [36]. 20% of the identified loci related to CAD are found near to genes that aid in the metabolism of LDL cholesterol, triglycerides, and Lp(a). 5-10% additional loci have been related to blood pressure. The nitric oxide signaling pathway also seems to protect against CAD [44].
The 9p21.3 locus
The alleles in the 9p21.3 locus are the most related to coronary atherosclerosis. These are present in 75% of the population (except in afro American people) [29]. The 9p21 locus is mainly associated with the first CAD event, instead of subsequent coronary events. Hereupon, it is suggested that this locus is more closely linked to coronary atherosclerosis than myocardial infarction (MI) [47].
Due to alternative splicing, a posttranscriptional process through which multiple distinct transcripts are produced from a single gene, the ANRIL (Antisense Noncoding RNA in the INK4 Locus) gene, also known as CDKN2BAS, can be formed into linear or circular transcripts. It is suggested that the linear form is atherogenic, while the circular form acts as atheroprotective. Consequently, CAD risk could depend on the ratio between circular and linear ANRIL [44]. The closest codifying genes correspond to CDKN2A and CDKN2B, which are involved in the cellular cycle regulation. Nevertheless, its effects on CAD still haven’t been unriddled, and more importantly, they are located at an important distance from the enhancer regions of the loci associated with CAD and CDKN2A/B [29]. The 9p21.3 locus is also related to abdominal and intracranial aneurysms, DM-2, metabolic syndrome, and stroke [48]. The pathophysiological mechanism of the locus still hasn’t been completely understood.
The 9p21 risk variant has been described in several ethnic groups, including Koreans, Japanese, Italians, and Hindi, with an insignificant contribution in the afro american population (Figure 3) [34]. Around 25% of all European people have 2 copies of the risk allele from the 9p21 locus and possess a 40% higher risk of CAD and a 2-fold risk for premature CAD [49]. To understand the differences between risk alleles from different populations, it is necessary to recall the concept of haplotype blocks, which are segments of DNA shared by population groups [49]. The European populations have more correlated SNP and larger haplotype blocks (≈20.7 kb) compared to the afro american race (≈8.8 kb). As a result, to create a genotype of an European population, fewer SNP are needed, in comparison to an African population, which has smaller haplotype blocks [50,51]. The majority of loci discovered in European populations have been reproduced on Asiatic populations, even though the odds ratio to some loci, including 9p21.3, have been less significant on Asiatics [52]. Secondly, most loci identified in the European population — including some polymorphisms on 9p21.3 —still haven't been replicated on African populations, despite the high prevalence of CAD and the smaller size of the haplotype blocks [49]. In the PAGE population study, it was reported that 25% of European SNP described by GWAS had significantly minor effects in African cohorts [53].
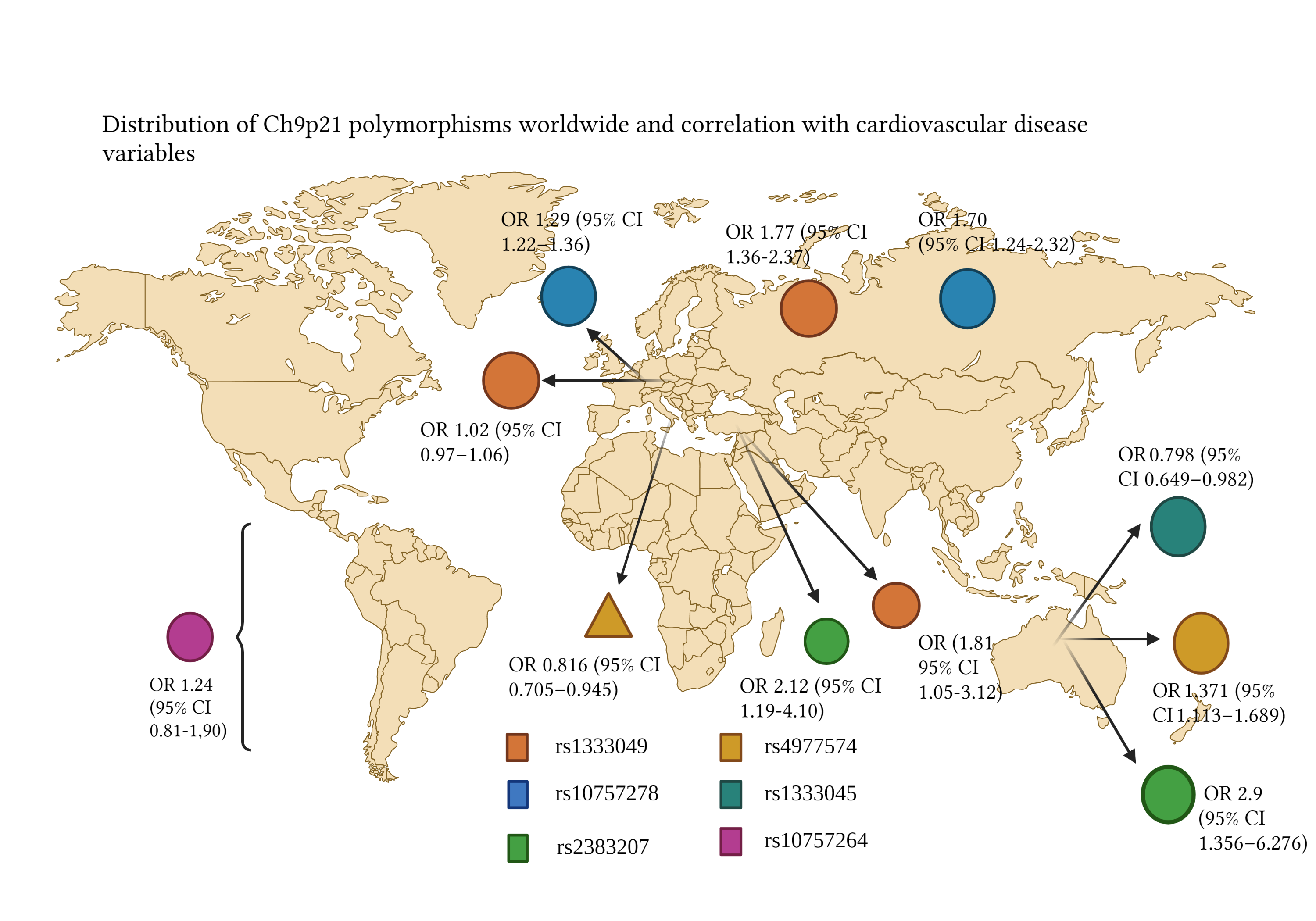
Figure 3. Correlation of Ch9p21 genotypes rs1333049 and rs10757278 with several CHD composites [57,58]
In the next paragraphs, we’ll describe some risk variants found in different populations.
Europeans
In the study by Patel R, et al. the GENIUS-CHD consortium was analyzed to examine the association between 9p21 locus variants (specifically rs1333049) and coronary artery disease. Besides, it compared the results with those of the CARDIoGRAMPlusC4D consortium. In total, 103357 people of European inheritance with established CAD, defined as prior MI and/or prior revascularization or angiography, were studied. In patients enrolled with an acute coronary syndrome, no evidence for a relationship between 9p21 genetic variation and CAD-related death or MI was found (OR, 1.02; 95% IC, 0.97–1.06), neither in patients with CAD and a prior MI (OR, 1.01; 95% IC, 0.96–1.05), nor those with CAD without prior MI (OR, 1.01; 95% IC, 0.95–1.08). A bigger association was established on women, patients with hypertension, and patients with kidney failure. Finally, a positive association was found with 9p21 locus and revascularization (OR, 1.07; 95% CI, 1.04–1.09) [54]. In the study by Zivotic I, et al. the relationship between the rs10757278 and rs518394 variants with a first non-fatal MI in patients with CAD was examined, with no observation of a relationship between the rs10757278 and rs518394 locus with a first non-fatal MI. Additionally, the association of rs518394 with MI and disease burden was not independent of other investigated risk factors [55].
It is worth noting that the rs10757278 polymorphism includes A and G alleles, among which A is the reference allele and G is the variant allele [56]. In this case, a bigger frequency of the risk allele G was described in control subjects. Besides, the association of rs518394 with MI is not independent of other risk factors. The existence of dysregulation in the genetic expression of chromosome 9p21.3 6 months after MI is suggested. Consequently, the rs10757278 allele represents a factor that favors the appearance of atherogenic complications [55]. In the meta-analysis by Chen G, et al. 12 case-control studies were gathered, to compare the G vs A allele of the rs10757278 polymorphism and the C vs G allele of rs1333049. An association between rs10757278 and MI was established (p=6.09 × 10−22, OR = 1.29, 95% IC 1.22–1.36). In doing this analysis by subgroups, no significant heterogeneity was found between the Asiatic and Caucasian populations. On the other hand, a strong relationship between rs10757278 and MI in the Asiatic (OR = 1.21, 95% CI 1.16–1.27) and the Caucasian population (OR =1.34, 95% CI 1.28–1.40) was established [56].
Russians
The study by Nikulina S, et al. aimed to study MI risk association with rs10757278 and rs1333049 polymorphisms. Regarding rs1333049, G and C alleles have been described, where C is the reference allele and G is the variant allele [56]. It was found that the MI risk was higher in people with a homozygous rs1333049 CC genotype, in comparison with a CG heterozygous genotype (1.77 vs 1.58 OR), and also a higher risk with homozygous rs10757278 GG genotype, compared with the CG heterozygous genotype (1.70 vs 1.36). Moreover, thanks to a regression model, it was concluded that the rs1333049 CC genotype was an independent predictive factor for MI (OR 1.71; 95% CI: 1.16-2.52). It was documented that the C allele of rs1333049 in patients without percutaneous coronary intervention (PCI), is linked to a greater risk of recurrent CAD, noting an odds ratio for the probability of recurrent acute coronary syndrome of 4.91 (95% CI: 1.45-16.66), a year after MI [57]. A higher survival ratio was observed in GG homozygotes (19.2 months) compared to CC homozygotes (18.5 months) in the PCI group, while the study group without PCI showed a survival rate of 16.2 months in CC homozygotes and 21.9 months in GG homozygotes. Thanks to this study, the influence of coronary intervention in the usage of genetic markers during hospitalization was documented. Thus, the use of chromosome 9p21 SNP is more reasonable during secondary intervention than during primary intervention [57]. The genotypification of chromosome 9p21.3 has clinical utility to predict complications after MI managed with PCI. This study proves that the presence or absence of coronary intervention during hospitalization must be taken into consideration in the evaluation of genetic markers as predictors of disease [57].
Italians
In the study by Pignataro P, et al. the objective was to verify the association between the rs1333049 C/G polymorphism from chromosome 9p21.3 and CAD in an Italian population. Altogether, 711 patients with CAD and 755 healthy subjects in the control group were studied. The main result was a relationship between the G allele and a lower risk of CAD (OR = 0.816; 95% IC [0.705–0.945]; p = 0.0065). No correlation was documented between rs1333049 and mortality or cardiovascular events [48].
Turkish
In the study by Çakmak HA, et al. 220 patients with CAD (67.7% male) and 240 individuals in the control group without CAD (39.5% male) were studied. The rs2383207 and rs1333049 polymorphisms were examined. They found that the GG genotype from rs1333049 is associated with greater CAD risk in the Turkish population (OR 1.81, 95% CI 1.05-3.12), contrasting with other studies, where the risk genotype had been CC [54,56-58]. Similarly, in this population, the A allele of rs2383207 strengthened the risk of CAD (OR 2.12, 95% CI 1.19-4.10), unlike other studies, where G was the risk allele. The AA variant of rs2383207 heightens the risk of CAD with an OR of 2.34; in contrast, the GG genotype of rs1333049 raises the risk with an OR of 2.32 (95% Cl 1.05-5.12) [58].
Chinese
The study of Hua L, et al. aimed to describe the correlation between polymorphisms rs4977574 (A > G) and rs1333045 (C > T) with CAD in a Chinese population. The main conclusions of the study were that the G allele of rs4977574 (p 0.003, OR 1.371, 95% CI 1.113 – 1.689) and the C allele of rs1333045 (p 0.035, OR 0.798, 95% CI 0.649 - 0.982) are sites of susceptibility for CAD. On the other hand, the T allele of rs1333045 can reduce the incidence of coronary events (p 0.035, OR 0.798, 95% CI 0.649–0.982). Thus, the A and T alleles may function as protecting factors against CAD [59]. Regarding another study, by Tang, O, the correlation between SNP rs4977574 and the development of CAD was examined in a Chinese population in the Province of Zhejiang, through the analysis of CT, CC, and TT polymorphisms. (60). Such polymorphisms had already been confirmed in Canadian and European populations [34,61,62]. The CC genotype was associated with a lower index of HbA1c. Furthermore, the presence of the C allele was associated with a reduced incidence of CAD. As a consequence, it is suggested that this polymorphism influences the susceptibility of CAD through glycemic level modifications [60]. It has been mentioned that the genetic factors contribute to 50% of susceptibility of DM-2 related to CAD [11-13, 60]. Finally, in the study by Jing J, Su L, Zeng Y, et al. a significant association between patients with 2-3 vessel disease and rs2383207 was observed; those with the GG allele had a higher probability of 3-vessel disease (OR: 2.9; 95% CI 1.356–6.276). Moreover, the presence of rs2383206 was also associated with CAD; those with GG genotype correspond to the highest grade of stenosis [63].
Latin America
Gong Y, et al. conducted a study that analyzed the association between 153 SNP in chromosome region 9p21 with a sample of case-controls (n=1460) and 2 SNPs (rs2383207 and rs10757278). Inside the cohort of INVEST-GENES, 8 SNP of statistical relevance were found, of which none were found of significance in the Hispanic and afro american population. On the other hand, only 1 SNP in the Hispanic population came close to statistical significance (rs10757264 with an OR 1.24, 95% IC 0.81-1,90). Thus, in this study, no SNP reached statistical relevance in the studied Hispanic population (n=390). [64]. This may be due to a greater level of genetic diversity, extensive population substructure, and lower linkage disequilibrium (the nonrandom association of alleles at different loci) in the Hispanic population, similar to that of the afro american population [50,51]. The evaluation of the same polymorphisms in different ethnic groups could allow the identification of pathologically important SNP. In this study, no relevant SNP in association with CAD was found in the Hispanic population, mainly from Puerto Rico. It is suggested that environmental factors have greater relevance or differences in the penetrance of genetic factors [66].
It is noteworthy that, despite the high prevalence of CAD in the Latin American population, there is a lack of studies that address the identification of risk variants in this group of patients.
Initially, the description of monogenic models of CAD (familial hypercholesterolemia) represented a critical point in the understanding of the role of genetics in complex diseases. Thanks to the recent implementation of GWAS studies, it has been possible to study which are the gene variants most associated with the development of CAD in several populations. Such studies have shown a wide heterogeneity, with outstanding differences between different population groups. Recently, the relevance of the 9p21 locus has been highlighted in several populations. Although the mechanisms through which it participates in CAD are not completely understood, future studies may aid in the implementation of polymorphisms as risk factors, with the possibility of identification of higher-risk patients and earlier therapeutic management, aiming at a reduction of CAD-related events. There is a lack of studies in the Latin American population, thus representing a promising field of research, given the high prevalence of CAD in this population group.
Figures created with BioRender.com
- Malakar AK, Choudhury D, Halder B, Paul P, Uddin A, et al. (2019) A review on coronary artery disease, its risk factors, and therapeutics. J Cell Physiol 234: 16812-16823. [Crossref]
- Khera AV, Kathiresan S (2017) Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet 18: 331. [Crossref]
- Khan MA, Hashim MJ, Mustafa H, Baniyas MY, Al Suwaidi SKBM, et al. (2020) Global Epidemiology of Ischemic Heart Disease: Results from the Global Burden of Disease Study. Cureus 12: e9349. [Crossref]
- Gosmanova EO, Mikkelsen MK, Molnar MZ, Lu JL, Yessayan LT, et al. (2016) Association of systolic blood pressure variability with mortality, coronary heart disease, stroke, and renal disease. J Am Coll Cardiol 68: 1375-1386 [Crossref]
- Mancia G, Messerli F, Bakris G, Zhou Q, Champion A, et al. (2007) Pepine Blood pressure control and improved cardiovascular outcomes in the International Verapamil SR-Trandolapril Study. Hypertension 50: 299-305. [Crossref]
- Mezue K, Goyal A, Pressman GS, Matthew R, Horrow JC, et al. (2018) Blood pressure variability predicts adverse events and cardiovascular outcomes in SPRINT. J Clin Hypertens 20: 247-1252. [Crossref]
- Wright JT, Bakris G, Greene T, SPRINT Research Group, Wright JT, et al. (2015) A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 373: 2103-2116.
- Gosmanov AR, Lu JL, Sumida K, Potukuchi PK, Rhee CM, et al. Synergistic association of combined glycemic and blood pressure level with risk of complications in US veterans with diabetes. J Hypertens 34: 907-913. [Crossref]
- Kovesdy CP, Alrifai AAZ, Gosmanova EO, Lu JL, Canada RB, et al. (2016) Age and outcomes associated with blood pressure in patients with incident CKD Clin J Am Soc Nephrol 11: 821-831 [Crossref]
- Luk AO, Ma RC, Lau ES, Yang X, Lau WWY, et al. (2013) Risk association of HbA1c variability with chronic kidney disease and cardiovascular disease in type 2 diabetes: prospective analysis of the Hong Kong Diabetes Registry. Diabetes Metab Res Rev 9: 384-390 [Crossref]
- Gorst C, Kwok CS, Aslam S, Buchan I, Kontopantelis E, et al. (2015) Long-term glycemic variability and risk of adverse outcomes: a systematic review and meta-analysis. Diabetes Care 38: 2354-2369. [Crossref]
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HAW (2008) 10-Year Follow-up of Intensive Glucose Control in Type 2 Diabetes. New England Journal of Medicine 359: 1577-1589.
- Hackshaw A, Morris JK, Boniface S, Tang JL, Milenkovic D (2018) Low cigarette consumption and risk of coronary heart disease and stroke: meta-analysis of 141 cohort studies in 55 study reports. BMJ 360: j5855. [Crossref]
- Steenaard RV, Ligthart S, Stolk L (2015) Tobacco smoking is associated with methylation of genes related to coronary artery disease. Clin Epigenet 7: 54.
- S. Yusuf, S. Hawken, S. Ounpuu, Dans T, Avezum A, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 364: 937-952. [Crossref]
- Magnani JW, Norby FL, Agarwal SK, Soliman EZ, Chen LY, et al. (2016) Racial Differences in Atrial Fibrillation-Related Cardiovascular Disease and Mortality: The Atherosclerosis Risk in Communities (ARIC) Study. JAMA Cardiol 1: 433-441. [Crossref]
- Anurag Mehta, Salim S. Virani, Colby R. Ayers, Wensheng Sun, Ron C. et al. (2020) Lipoprotein(a) and Family History Predict Cardiovascular Disease Risk. J Am Coll Cardiol 76: 781-793. [Crossref]
- Akin I, Nienaber CA (2015) "Obesity paradox" in coronary artery disease. World J Cardiol 7: 603-608. [Crossref]
- Garcia-Labbé D, Ruka E, Bertrand OF, Voisine P, Costerousse O, et al. (2015) Obesity and coronary artery disease: evaluation and treatment. Can J Cardiol 31: 184-94 [Crossref]
- Ye J (2013) Mechanisms of insulin resistance in obesity. Front Med 7: 14-24. [Crossref]
- Bangalore S, Fayyad R, Laskey R, DeMicco DA, Messerli FH, et al. (2017) Body-Weight Fluctuations and Outcomes in Coronary Disease. New England Journal of Medicine 376: 1332-1340.
- Whitlock G, Lewington S, Sherliker P, Clarke R, Emberson J, et al. (2009) Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet 373: 1083-1096. [Crossref]
- Pasternak RC (2003) Report of the adult treatment panel III: the 2001 National Cholesterol Education Program guidelines on the detection, evaluation and treatment of elevated cholesterol in adults. Cardiol Clin 21: 393-398. [Crossref]
- Peter Alagona, Tariq Ali Ahmad, Cardiovascular Disease Risk Assessment and Prevention: Current Guidelines and Limitations. Med Clin North Am 99: 711-731, [Crossref]
- Goff DC, Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB, et al. (2013) 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 129: S49-S73. [Crossref]
- González C, Rodilla E, Costa JA, Justicia J, Pascual JM (2006) Comparación entre el algoritmo de Framingham y el de SCORE en el cálculo del riesgo cardiovascular en sujetos de 40-65 años. Medicina Clínica 126: 527–531.
- Rotter JI, Lin HJ (2020) An Outbreak of Polygenic Scores for Coronary Artery Disease. J Am Coll Cardiol 75: 2781-2784. [Crossref]
- Aragam KG, Natarajan P (2020) Polygenic Scores to Assess Atherosclerotic Cardiovascular Disease Risk. Circ Res 126: 1159-1111. [Crossref]
- Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, et al. (2007) WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 357: 443-453. [Crossref]
- Zuk O, Hechter E, Sunyaev SR, Lander ES (2012) The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 109: 1193-1198.
- Musunuru K, Hershberger RE, Day SM, Klinedinst NJ, Landstrom AP, et al. (2020) Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ Genom Precis Med 13: e000067. [Crossref]
- Altshuler D, Daly MJ, Lander ES (2008) Genetic mapping in human disease. Science 322: 881-888. [Crossref]
- Risch N, Merikangas K (1996) The Future of Genetic Studies of Complex Human Diseases. Science 273: 1516-1517. [Crossref]
- Roberts R (2014) Genetics of Coronary Artery Disease. Circ Res 114: 1890-1903. [Crossref]
- Crouch DJM, Bodmer WF (2020) Polygenic inheritance, GWAS, polygenic risk scores, and the search for functional variants. Proc Natl Acad Sci U S A 117: 18924-18933. [Crossref]
- Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, et al. (2015) A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 47: 1121-1130. [Crossref]
- Kathiresan S, Srivastava D (2012) Genetics of human cardiovascular disease. Cell 148: 1242-1257. [Crossref]
- Fahed AC, Nemer GM (2011) Familial Hypercholesterolemia: The Lipids or the Genes? Nutr Metab (Lond) 8: 23. [Crossref]
- Defesche J, Gidding S, Harada-Shiba M, Hegele RA, Santos RD, et al. (2017) Familial hypercholesterolaemia. Nat Rev Dis Primers 3: 17093. [Crossref]
- Strachan T, Andrew P, Strachan T (2011) Human Molecular Genetics. New York: Garland Science,
- Barth AS, Tomaselli GF (2016) Gene scanning and heart attack risk. Trends Cardiovasc Med 26: 260-265. [Crossref]
- Bouhairie VE, Goldberg AC (2015) Familial Hypercholesterolemia. Cardiol Clin 33: 169-179. [Crossref]
- Benito-Vicente A, Uribe K, Jebari S, Galicia-Garcia U, Ostolaza H, et al. (2018) Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. Int J Mol Sci 34: 526–530. [Crossref]
- Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, et al. (2016) Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J Am Coll Cardiol 67: 2578-2589. [Crossref]
- Safarova MS, Kullo IJ (2016) My Approach to the Patient With Familial Hypercholesterolemia. Mayo Clin Proc 91: 770-786. [Crossref]
- Müller C (1938) Xanthomata, hypercholesterolemia, angina pectoris. Acta Med Scand 89: 75-84.
- Hernesniemi JA, Lyytikäinen LP, Oksala N, Seppälä I, Kleber ME, et al. (2015) Predicting sudden cardiac death using common genetic risk variants for coronary artery disease. Eur Heart J 36: 1669-1675. [Crossref]
- Pignataro P, Pezone L, Di Gioia G, Franco D, Iaccarino G, et al. (2017) Association Study Between Coronary Artery Disease and rs1333049 Polymorphism at 9p21.3 Locus in Italian Population. J of Cardiovasc Trans Res 10: 455-458. [Crossref]
- Ruth M, Anne TH (2016) Genetics of Coronary Artery Disease. Circ Res 118: 564-578. [Crossref]
- Campbell MC, Tishkoff SA (2008) African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet 9: 403-433. [Crossref]
- Edwards SL, Beesley J, French JD, Dunning AM (2013) Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet 93: 779-797. [Crossref]
- Marigorta UM, Navarro A (2013) High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet 9: e1003566. [Crossref]
- Carlson CS, Matise TC, North KE, Haiman CA, Fesinmeyer MD, et al. (2013) PAGE Consortium. Generalization and dilution of association results from European GWAS in populations of non-European ancestry: the PAGE study. PLoS Biol 11: e1001661. [Crossref]
- Patel RS, Asselbergs FW, Quyyumi AA, Palmer TM,Finan CI, et al. (2014) Genetic variants at chromosome 9p21 and risk of first versus subsequent coronary heart disease events: a systematic review and meta-analysis. J Am Coll Cardiol 63: 2234-2245. [Crossref]
- Zivotic I, Djuric T, Stankovic A, Milasinovic D, Stankovic G, et al. (2019) CDKN2B gene expression is affected by 9p21.3 rs10757278 in CAD patients, six months after the MI. Clin Biochem 73: 70-76. [Crossref]
- Chen G, Fu X, Wang G, Liu G, Bai X (2015) Genetic Variant rs10757278 on Chromosome 9p21 Contributes to Myocardial Infarction Susceptibility. Int J Mol Sci 16: 11678–11688. [Crossref]
- Nikulina S, Artyukhov I, Shesternya P, Gavrilyuk O, Maksimov V, et al. (2019) Clinical application of chromosome 9p21.3 genotyping in patients with coronary artery disease. Exp Ther Med 18: 3100-3108. [Crossref]
- Çakmak HA, Bayoglu B, Durmaz E, Can G, Karadag B, et al. (2015) Evaluation of association between common genetic variants on chromosome 9p21 and coronary artery disease in Turkish population. Anatol J Cardiol 15: 196-203. [Crossref]
- Hua L, Yuan JX, He S, Zhao C, Jia Q, et al. (2020) Analysis on the polymorphisms of site RS4977574, and RS1333045 in region 9p21 and the susceptibility of coronary heart disease in Chinese population. BMC Med Genet 21: 36. [Crossref]
- Tang O, Lv J, Cheng Y, Qin F (2018) he Correlation Between 9p21 Chromosome rs4977574 Polymorphism Genotypes and the Development of Coronary Artery Heart Disease. Cardiovasc Toxicol 17: 185-189. [Crossref]
- Munir MS, Wang Z, Alahdab F, Steffen MW, Erwin PJ, et al. (2014) The association of 9p21-3 locus with coronary atherosclerosis: A systematic review and meta-analysis. BMC Med Genet 15: 66. [Crossref]
- Rivera NV, Carreras-Torres R, Roncarati R, Viviani-Anselmi C, De Micco F, et al. (2013) Assessment of the 9p21.3 locus in severity of coronary artery disease in the presence and absence of type 2 diabetes. BMC Med Genet 14: 11. [Crossref]
- Jing J, Su L, Zeng Y, Tang X, Wei J, et al. (2016) Variants in 9p21 Predicts Severity of Coronary Artery Disease in a Chinese Han Population. Ann Hum Genet 80: 274-281. [Crossref]
- Gong Y, Beitelshees AL, Cooper-DeHoff RM, Lobmeyer MT, Langaee TY, et al. (2011) Chromosome 9p21 haplotypes and prognosis in white and black patients with coronary artery disease. Circ Cardiovasc Genet 4: 169-178. [Crossref]
- Slatkin M (2008) Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9: 477-485.
- Dandona S, Stewart AF, Chen L, Williams K, So D, et al. (2010) Gene dosage of the common variant 9p21 predicts severity of coronary artery disease. J Am Coll Cardiol 56: 479-486. [Crossref]
Editorial Information
Editor-in-Chief
Akira Sugawara
Tohoku University Graduate School of Medicine
Article Type
Review Article
Publication history
Received date: December 09, 2021
Accepted date: December 20, 2021
Published date: December 23, 2021
Copyright
©2021 Arce-Sandoval CR. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation
Arce-Sandoval CR, Zavala-Romero L, Romero-Montiel RE, Asch-Abadi S, Gurrola-Luna H, et al. (2021 Genetics of coronary artery disease: State of the art in the last decade. Clin Res Trials 7: doi: 10.15761/CRT.1000360