Abstract
Hereditary spastic paraparesis is a group of diseases that have as their main clinical aspect weakness and spasticity in the lower limbs, associated with a large number of genes, but involving a more restricted number of cellular functions that cause dysfunction of the neurons that make up the corticospinal tract. We will present a series of 3 cases of our service, mainly addressing the relation between the neuroimaging findings and the clinical alterations observed, with later literature review, showing the importance of this relationship, since not all services have a molecular diagnostic service when the acquired causes are removed.
Introduction
Hereditary Spastic Paraplegia (PEH) is a clinical entity that involves a heterogeneous group of genetic diseases related to retrograde degeneration of the corticospinal tract, which presents mainly cardinal changes of weakness and spasticity of lower limbs.
Currently there are at least 60 types of genetic changes related to the disease. Large series of cases in developed countries record about 70% of cases with autosomal dominant pattern. This pattern usually presents in the pure form, that is to say, with predominant spastic paraparesis, being able to be associated with vesical alterations, slight cognitive decline, cavus feet and alterations of appendicular distal sensibility. The most common mutation in these cases is SPG4.
The other cases present an autosomal recessive or X-linked pattern, being associated with the so-called complicated form of the disease, which usually presents with several neurological manifestations resulting from the involvement of several, long or non-dependent treatments. The main neurological manifestations presented together are: amyotrophy, optic atrophy, pigmentary retinopathy, dementia, parkinsonism, deafness, peripheral neuropathy and epilepsy. The mutation most related to these cases is SPG11, which is also the most related to neuroimaging characteristic alterations, such as fine corpus callosum and signs of the lynx ears.
The pathophysiology of the disease is still not well understood, but it is believed that the genetic alterations are related to dysfunction of the axoplasmic flow of the corticospinal tract through molecular changes in neuronal cytoskeletal structures responsible for membrane trafficking and axonal transport of vesicles and endosomes [1].
Case 1
M.R.S, female, 46 years old, started difficulties for ambulation around 36 years of age. About three years later, she also presented voiding urgency and occasional episodes of urinary incontinence.
At the physical examination, the patient presented with paresthesia, muscular strength grade III, with significant spasticity and exalted osteotendinous reflexes, without clones, with signs of Babinski and Hoffmann present bilaterally. She reported a family history of similar symptoms started in a daughter at age 5, a sister at age 48, and a nephew (son of this sister) at age 15. Brain MRI was normal (Figures 1 and 2).
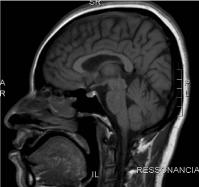
Figure 1. Brain MRI: Sagittal T1: no significant changes.
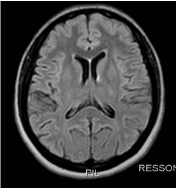
Figure 2. Axial T2-FLAIR: no significant changes.
Case 2
D.S.S, male, 30 years of age, followed from age 15, due to tremor in the left upper limb, progression to the right upper limb after one year, and presented partial response to levodopa therapy. He presented mild cognitive disorder, with reports of early school difficulties. He began to present psychomotor retardation evident at around 10 years of age.
At age 22, he was submitted to MRI of the brain, showing signs of diffuse tapering of the corpus callosum (Figures 3 and 4), and a slight diffuse alteration of the sign of periventricular deep white matter, and could not be correlated at the time with the clinical. At age 24, he developed weakness of the lower limbs, progressive, with difficulty climbing stairs and reporting frequent falls.

Figure 3. Brain MRI: Sagital T1: signs of diffuse tapering of the corpus callosum.
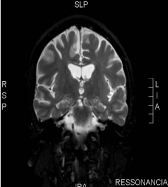
Figure 4. Coronal T2: tenuous diffuse alteration of sign of periventricular deep white matter.
Bilateral spasticity of the lower limbs, with symmetrical and exalted osteotendinous reflexes, bilaterally depleted clusters, and spastic gait were identified in an outpatient follow-up. He presented with dysarthria, dysdiadochokinesia and moderate cognitive decline. Family history without relevance.
Case 3
R.P.O, male, 26 years old, started seizures at 7 years of age, and at the age of 9 he developed generalized tonic-clonic seizures, using valproic acid and phenobarbital. He started talking only at 3 years of age, and always had difficulty accompanying the school, being evaluated with mild cognitive impairment. At the age of 13, he began to struggle for ambulation, slow progression, and worse worsening after 20 years of age.
He had physical examination, spasticity, and exalted reflexes in lower limbs. At 18 he stopped presenting epileptic seizures, and the anticonvulsant medications were successfully withdrawn. Family history without relevance. MRI showed tapering of the anterior portion of the body and knee of the corpus callosum and hypersignal in the T2 / FLAIR sequences with a triangular aspect adjacent to the frontal horns of the lateral ventricles (Figures 5 and 6).
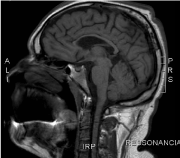
Figure 5. Brain MRI: Sagittal T1: tapering of the anterior portion of the body and knee of the corpus callosum
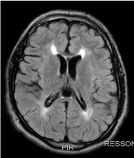
Figure 6. Axial T2-FLAIR: hypersignal in the T2 / FLAIR sequences with triangular aspect adjacent to the frontal horns of the lateral ventricles (sign of the " ears of lynx")
Discussion
All the cases presented are compatible with the syndromic diagnosis of Hereditary Spastic Paraplegia. When the pure clinical picture is accompanied by the family history compatible with autosomal dominant pattern, as in case 1, the diagnosis is evident. In these cases PEH SPG4 is the most prevalent, accounting for about half of the cases, especially when the condition is present in adulthood [2,3]. However, the recessive pattern usually presents with no relevant family history.
Diagnosis requires exclusion of other causes, and the investigation is directed according to the form and age of presentation of the case, since the genetic diagnosis is inaccessible to most services.
In relation to case 2, it can be considered an etiological diagnosis of PEH associated with the SPG21 gene, or Mast's syndrome, which relates the presentation to early adulthood, associated with the finding of a thin corpus callosum neuroimaging, cognitive decline, alterations extrapyramidal and cerebellar alterations.
Another possibility would be PEH SPG11, which usually presents in childhood, with cognitive decline and peripheral neuropathy. We can consider this as the probable alteration of case 3, by the age of compatible presentation and the most specific finding to neuroimaging, called signal of the lynx ears [4,5].
Definitive diagnosis requires genetic testing, which is generally inaccessible in most neurology services, even at referral centers. In these cases, the clinical diagnosis is imposed after the vascular, infectious, toxic, autoimmune, metabolic, inflammatory and neoplastic differential diagnoses are excluded.
Brain MRI, however, may help in the etiological diagnosis, evidencing relatively specific alterations within the clinical and evolutionary context, related to some forms of PEH.
Currently the treatment is based on multidisciplinary neurofunctional rehabilitation and symptomatic clinical control of neurological complications. The development of biomolecular techniques and the better understanding of this clinical entity may, in the future, provide targeted therapies and consequently more effective for this group of diseases.
The availability of genetic testing in reference services in a broader way can help in the identification of new cases, new phenotypes and perhaps a better understanding of the natural history of the disease, ways of inheritance and thus better select the patients suitable for therapeutic proposals that may to emerge [6,7].
References
- Salinas S, Proukaki2021 Copyright OAT. All rights reservitary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet neurol 7: 1127-1138. [Crossref]
- Faber I, Servelhere KR, Martinez AR, D'Abreu A, Lopes-Cendes I, et al. (2014) Clinical features and management of hereditary spastic paraplegia. Arq Neuropsiquiatr 72: 219-226. [Crossref]
- França Jr MC, Dogini DB, D'Abreu A, Teive HA, Munhoz RP, et al. (2014) SPG4-related hereditary spastic paraplegia: frequency and mutation spectrum in Brazil. Clin Genet 86: 194-196. [Crossref]
- Kang SY, Lee MH, Lee SK, Sohn YH (2004) Levodopa-responsive parkinsonism in hereditary spastic paraplegia with thin corpus callosum. Parkinsonism Relat Disord 10: 425-427. [Crossref]
- Schule R, Schlipf N, Synofzik M, Klebe S, Klimpe S, et al. (2009) Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry 80: 1402-1404. [Crossref]
- Parodi L, Fenu S, Stevanin G, Durr A (2017) Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev Neurol (Paris) 173: 352-360. [Crossref]
- Souza PV, de Rezende Pinto WB, de Rezende Batistella GN, Bortholin T, Oliveira AS. Hereditary Spastic Paraplegia: Clinical and Genetic Hallmarks. Cerebellum 16: 525-551. [Crossref]