Hyperhemolysis syndrome (HS) is a serious and life-threatening complication of red blood cell (RBC) transfusion and has been well described in SCD patients. HS can be further classified into acute and delayed forms. Delayed form of HS occurs mainly by bystander mechanism in which, there may be evidence of complement deposition on autologous red cells, with a positive direct antiglobulin test (DAT) due to C3 persisting for up to 100 days. We aim to highlight the importance of recognizing episodes of HS as they present as pain crisis in SCD patients following transfusion and preventing wrong management of crisis with RBC transfusion which can increase hemolysis and may cause life-threatening anemia. Here we report cases of siblings (known cases of SCD) with bystander hemolysis, presenting 1 to 2 weeks after blood transfusion with pain crisis showing signs and symptoms of accelerated hemolysis. Both brother and sister had positive DAT with only C3d positivity, lack of autoantibody in the eluate and formation of new alloantibodies (anti-E in sister and anti-E with anti-c in brother). Both the cases were successfully managed with avoidance of further transfusions and administration of corticosteroids. Management of HS in SCD depends on the severity of the symptoms, ranging from supportive care to immunosuppression, and optimization of erythropoiesis. HS must be considered in any recently transfused SCD patient presenting with an acute pain crisis.
hyperhemolysis syndrome, bystander hemolysis, vaso-occlusive crisis, alloantibodies, sickle cell disease
Sickle cell anemia (SCA) is a genetic disorder characterized by homozygous hemoglobin S (Hb S), chronic haemolytic anemia and painful episodes. The hallmark of sickle cell disease is the vaso-occlusive pain crisis [1]. It is the most common clinical manifestation but can occur with varying frequency in different individuals. SCD patients need RBC transfusion for managing complications and reducing morbidity during surgery. HS is a serious and life-threatening complication of RBC transfusion and has been well described in SCD patients. HS may present with fever, pain crisis, development of severe anemia after transfusion, evidence of hemolysis (hemoglobinuria, hyperbilirubinemia and raised lactate dehydrogenase[LDH]) and a fall in absolute reticulocyte count [2-8].
HS can be further classified into acute and delayed forms [6,9]. Acute form usually occurs within 7 days of transfusion with a negative DAT and no new alloantibody formation whereas delayed form usually occurs after 7 days of transfusion with a positive DAT and new alloantibodies. Delayed form of HS occurs mainly by bystander mechanism in which, there may be evidence of complement deposition on autologous red cells, with a positive DAT due to C3 persisting for up to 100 days [10]. This phenomenon has been called “bystander hemolysis.” Typically, bystander hemolysis is mild and is differentiated from autoimmune hemolytic anemia (AIHA) by the absence of anti-IgG reactivity in the DAT and the lack of autoantibody in the eluate [11]. In the setting of SCD, bystander hemolysis can be very severe. It is important to realize that further transfusion in this setting may exacerbate the anemia and even prove fatal.
We aim to highlight the importance of recognizing such episodes as they present as pain crisis in SCD patients following transfusion and preventing wrong management of crisis with RBC transfusion which can increase hemolysis and may cause life-threatening anemia. We report cases of bystander hemolysis in siblingswith SCA, 1 to 2 weeks after blood transfusion.
A 13 years old girl known case of SCD was referred to our hospital with pain crisis from her native town, where she was treated with analgesics, intravenous fluids, and two units of ‘O’ Rh D positive whole blood transfusion. Pretransfusion reports from her native town showed hemoglobin (Hb) to be 8.2 gm%, Hematocrit (HCT) as 25% and bilirubin to be 2.7 mg/dl (unconjugated -1.9 mg/dl). After admission to the Paediatrics Department of our hospital, her blood test reports showed haemoglobin (Hb) level of 6.2 gm%, Hematocrit (HCT) of 20%, LDH of 943 U/L and Bilirubin 6 mg/dl (unconjugated -5.5 mg/dl). She was then transfused with three units of ‘O’ Rh D positive packed red blood cells (PRBC) and was referred to our department after 10 days for low hemoglobin despite blood transfusions.
Her reports then showed Hb of 5 gm%, HCT of 15%, LDH of 1606 U/L, Bilirubin of 7.2 mg/dl (unconjugated-6.8 mg/dl) and reticulocyte 0.2%. She had signs and symptoms of accelerated hemolysis evidenced by an unexplained fall in Hb, elevated LDH, elevated bilirubin, and hemoglobinuria, all occurring between 1 to 2 weeks after RBC transfusion.
Immunohematological workup as per our departmental protocol was done. Her blood group was found to be ‘O’ Rh D positive by conventional tube technique. DAT was positive (3+) on Ortho Biovue polyspecific cassette. Monospecific DAT with Tulip antisera came to be C3d-positive (2+) and IgG-negative with a negative elute. Auto-control and Thermal amplitude test was negative at 37°C, 4°C and room temperature. Alloantibody anti-E was found after antibody screening and identification using Ortho Surgiscreen and Resolve Panel A (Figures 1 and 2).
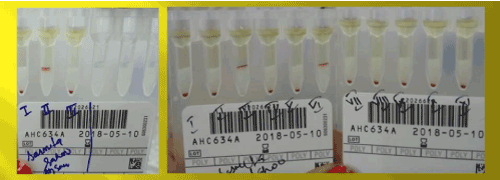
Figure 1. Antibody screening and identification of the sister using ortho surgiscreen and resolve panel A
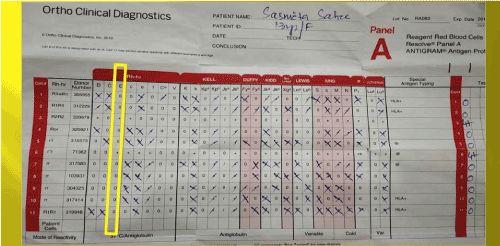
Figure 2. Antibody identification antigram of the sister; antibody identified anti-E
Considering the hypothesis of HS and bystander hemolysis, she was started with intravenous methylprednisolone. She was discharged after her Hb level gradually improved to 8 gm%.
Meanwhile, her brother, aged 14 years also a known case of SCD was admitted to our hospital with pain crisis. He too was referred to us for low Hb despite six blood transfusions at his native town. His Hb was decreased to 4 gm% from a pretransfusion value of 8 gm%. He too had signs and symptoms of accelerated hemolysis with unexplained fall in Hb, elevated LDH (1711 U/L), elevated bilirubin (8 mg/dl), reticulocyte 0.1% and hemoglobinuria.
Immunohematological workup showed blood group to be ‘O’ Rh D positive and DAT Positive (2+) with monospecificIgG negative and C3d positive (1+) and negative elute. Auto-control and thermal amplitude test were negative at 37°C,4°C and room temperature. Alloantibodies anti-E and anti-c were found in his serum after antibody screening and identification (Figures 3 and 4). He was also treated with intravenous methylprednisolone considering bystanders hemolysis. His clinical condition and Hb improved gradually after the treatment.
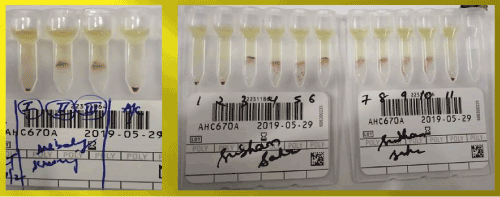
Figure 3. Antibody screening and identification of the brother using ortho surgiscreen and resolve panel A
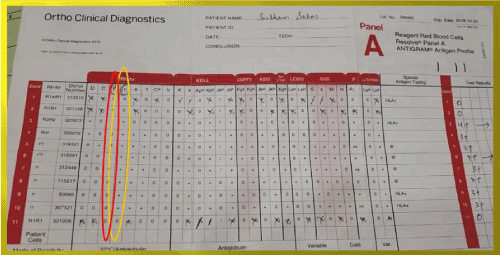
Figure 4. Antibody identification antigram of the brother; antibody identified anti-E and anti-C
Both brother and sister improved clinically after treatment with corticosteroid and avoidance of further transfusion. Blood reports of the sister after one month showed Hb of 10.3 gm%, HCT of 32.5% and DAT positive (1+). Hb of the brother also improved to 10.2 gm%, HCT to 31.2% after three weeks with a negative DAT. They are being followed up and there were no further crisis episodes in both of the siblings even after the completion of one year of the HS episode.
The pathogenesis of HS appears to be complex and remains unclear as the Hb level decreases below the pretransfusion level. Bystander hemolysis [8], suppression of erythropoiesis and RBSs being destroyed by activated macrophages [6] are the possible mechanisms of HS.
The term bystander hemolysis was first coined by Petz and Garraty to describe the phenomenon of “immune hemolysis of cells that are negative for antigen against which the relevant antibody is directed” [12]. Both brother and sister in our case had signs and symptoms of accelerated hemolysis evidenced by an unexplained fall in Hb, elevated LDH, elevated bilirubin, and hemoglobinuria, all occurring between 1 to 2 weeks after blood transfusion, which suggests HS by bystander mechanism. King et al. suggested the bystander hemolysis mechanism and his group has reported five cases of HS [8]. The authors concluded that the cells were destroyed by bystander hemolysis in two out of five cases.
As both brother and sister showed a post-transfusion Hb level lower than pretransfusion value, it suggested the destruction of both their own as well as transfused RBCs. This is unlike the conventional delayed hemolytic transfusion reactions (DHTRs) where an anamnestic immune response to antigen carried by the transfused cells constitutes a prerequisite. Persisting positive DAT in both of them due to C3d with an absence of anti-IgG reactivity in the DAT and the lack of autoantibody in the eluate made the diagnosis of AIHA unlikely.
Formation of new alloantibodies, anti-E in sister and anti-E with anti-c in the brother in the post-transfusion period justified the hypothesis of HS and bystander hemolysis. Darbi et al. have reported a delayed form of HS due to anti-K which represents a typical example of bystander hemolysis [13]. The patients experienced ongoing hemolysis for 9 weeks, despite receiving 19 units of K-crossmatched compatible units.
Absolute reticulocytosis is generally presented as a mechanism that is compensatory for hemolysis in steady-state SCD. However, in HS, a fall in reticulocyte count during hemolysis was recorded and recovery manifested by an increase in reticulocyte count. Both brother and sister in our case showed reticulocytopenia which made the diagnosis of HS very likely. Association of reticulocytopenia with HS was first described by Petz et al. [2]. They reported four delayed and one acute forms of HS and suggested that the apparent increased hemolysis of autologous RBC was partly due to transfusion induced suppression of erythropoiesis.
Awareness and diagnosis of HS very essential. It is important to realize that further transfusion in this setting may exacerbate the anemia and even prove fatal [2,13]. Management depends on the severity of anemia and the speed of hemolysis. In our case both brother and sister were successfully managed with avoidance of further transfusions and administration of corticosteroids.
HS is a serious and potentially life-threatening complication in SCD which is now been well described. So, awareness is necessary as without a high index of suspicion the case may go unrecognized as they present as pain crisis in SCD patients. Treatment with steroids and IVIG should be considered in severe life-threatening HS.
- Kenneth A, Lichtman Marshall A, William BE (2010) Hematology, 8thed: McGraw-Hill, Chepes (La Rioja), Argentina, pp: 881-882.
- Petz LD, Calhoun L, Shulman IA, Johnson C, Herron RM (1997) The sickle cell hemolytic transfusion reaction syndrome. Transfusion 37: 382-392. [Crossref]
- Cullis JO, Win N, Dudley JM, Kaye T (1995) Post-transfusion hyperhaemolysis in a patient with sickle cell disease: use of steroids and intravenous immunoglobulin to prevent further red cell destruction. VoxSanguinis 69: 355-357.
- Win N, Doughty H, Telfer P, Wild BJ, Pearson TC (2001) Hyperhemolytic transfusion reaction in sickle cell disease. Transfusion 41: 323-328.
- Win N, Sinha S, Lee E, Mills W (2010) Treatmentwith intravenous immunoglobulin and steroids may correct severe anemia in hyperhemolytic transfusion reactions: case report and literature review. Transfusion Medicine Reviews 24: 64-67.
- Win N, Doughty H, Telfer P, Wild B, Pearson T (2001) Hyperhaemolytic transfusion reaction in sickle cell disease. Transfusion 41: 323-328.
- Talano JA, Hillery CA, Gottschall JL, Baylerian DM, Scott JP (2003) Delayedhemolytic transfusion reaction/hyperhemolysis syndrome in children with sickle cell disease. Pediatrics 111: e661-e665.
- King KE, Shirey RS, Lankiewicz MW (1997) Simultaneous occurrence of posttransfusionpurpura due to anti-HPA-1a and a delayed transfusion reaction due to anti-Jk(b). Transfusion 37: 449-450.
- Win N (2004) Blood transfusion therapy for hemoglobinopathies. In: Practical Management of Haemoglobinopathies. Okpala I(Ed.). Blackwell Publishing, Oxford, UK, pp: 99-106.
- Salama A, Mueller-Eckhardt C (1984) Delayed haemolytic transfusion reactions. Evidence for complement activation involving allogenic and autologous red cells. Transfusion 24: 188-193.
- Popovsky Mark A (2012) Transfusion Reactions. 4thed: AABB, Bethesda, Maryland, pp: 6-7.
- Petz LD (2006) Bystander immune cytolysis. Transfusion Medicine Reviews 20: 110-140.
- Darabi K, Dzik S (2005) Hyperhaemolysis syndrome in anemia of chronic disease. Transfusion 45: 1930-1933.