Hypertensive heart failure (HHF) is a major public health problem associated with significant morbidity and mortality. Its cardinal characteristics are left ventricular hypertrophy and diastolic dysfunction resulting from the response to biochemical stress imposed on the left ventricle (LV) by a chronic and progressive increase in blood pressure. However, the precise understanding of the nature of HHF has been partial because of its many different terminologies and definitions making comparison and aggregation of studies challenging. In addition, the current heart failure (HF) classification systems and clinical guidelines based on morphological and/or functional characteristics do not inspire research into etiology-specific classification and treatment. However, with risk factors for the development of HHF such as obesity, diabetes, sedentarism, smoking and high salt intake increasing, there is need to improve the diagnosis and clinical management of HHF. The present review thus aggregates available research evidence on HHF to provide a comprehensive understanding of its clinical status.
hypertensive cardiomyopathy, hypertensive cardiopathy, hypertensive heart disease, hypertensive heart failure
Heart failure (HF) is a cardiogeriatric disease carrying a high burden of comorbidity, symptoms, suffering and death thus requires optimal medical care. However, even with increasing use of evidence-based and guideline-recommended medical care, hospitalization and mortality rates for HF patients have remained unacceptably high [1]. The biology of HF is complex and diverse posing challenges to the development of efficacious therapies. The inadequacy of the current HF therapies developed from objective evaluation of functional and structural status of the heart indicate the need for specialized interventions or a fundamental change in therapeutic approach [2]. The need has motivated research into the development of etiology-specific therapies targeted to improve drug development and drug response monitoring [3]. The motivation has influenced the expansion of HF research from the traditional systolic versus diastolic classification to include etiology-based HF phenotypes particularly HF in the setting of hypertension and ischemia [4]. Hypertensive HF (HHF) is a recently described etiology-based HF phenotype caused by continuous cardiac insult by chronic hypertensive heart disease (HHD) but potentially treatable by anti-hypertensive medication. Although over the years the literature on the various facets of HHF has been accumulating, the risk and mechanism of HHF has been partially demonstrated. Moreover, due to the common co-existence of coronary artery disease (CAD) and hypertension in HF population, their individual contribution to the pathophysiology of HF has been difficult to disentangle [4]. In the present article, we review published evidence to provide an overview of the salient aspects of HHF from a clinical standpoint.
The terminology of HF secondary to hypertension varies widely limiting its definitional uniformity. Cardiology societies and scholars have variously termed this phenotype of HF as hypertensive HF [4], hypertensive cardiomyopathy [5], hypertensive cardiopathy [6] or hypertensive heart disease (HHD) [7-9]. The initial definition of HHF by the New York Heart Association (NYHA) made in 1979 was an anatomofunctional alteration characterized by left ventricular (LV) hypertrophy (LVH) and cardiac failure in patients with systemic hypertension [6]. The definition does not distinguish HHF from HF. Recent definitions have also linked HHF to LVH. HHF is a form of decompensated HF usually characterized by complex myocardial alterations including disruptions in cardiac function and electric activity, and/or coronary flow abnormalities resulting from the response to biochemical stress imposed on the left ventricle (LV) by a chronic and progressive increase in blood pressure (BP) [4]. Its cardinal clinical manifestation is the presence of LV hypertrophy (LVH) in the absence of any other cause but arterial hypertension [7]. It has also been defined as a structural cardiac disorder generally accompanied by concentric LVH associated with diastolic or systolic dysfunction in patients with persistent systemic hypertension occurring in the absence of any other cardiac disease capable of causing myocardial hypertrophy or cardiac dysfunction such as hypertrophic cardiomyopathy, Fabry disease or cardiac amyloidosis [5].
Although these definitions are accurate, they seem incomplete because they are difficult to apply in clinical practice. In addition, while LVH is the most typical cardinal complication in HHF patients, it is not a distinguishing characteristic of HHF as well as hemodynamic mechanisms are not the only ones involved in the development of LVH [5]. Many patients may complain, especially at the onset of the disease, of other symptoms of heart involvement, and at advanced stages, arrhythmias and coronary ischemia could exacerbate symptoms [4,5]. The Working Group of the Spanish Society of Cardiology (SSC) [6] proposes that a clinically meaningful definition of HHF should include myocardial alterations induced by (a) a chronic blood pressure (BP) elevation; (b) alterations to the left ventricle (LV); (c) myocardial ischemia (MI); and (d) atrial fibrillation, which are the most frequent cardiac complications in hypertensive patients. Such a clinical definition would potentially improve risk stratification and guide treatment [6]. However, despite a good number of studies performed over the years, no clinically meaningful definition of HHF is available. Furthermore, the 2013 European Society of Hypertension (ESH) and European Society of Cardiology (ESC) is non-specific to HHF, only providing definition and management of arterial hypertension and the associated increased risk in CVD [10]. The SSC proposes HHF should describe a complex and variable syndrome that usually should but not necessarily include clinical manifestations secondary to LVH and LV diastolic or systolic dysfunction, MI and rhythm abnormalities derived from myocardial response to chronically elevated BP [6].
Classification of HHF is clinically significant to categorize patients who are at the greatest risk of HF as well as to guide the selection of the best available treatment strategy. For a long time, HHF guidelines focused on BP cut-off values as the only or the primary variables determining the need for and type of therapy. The implication is that HHF currently lacks a well-established and specific classification for therapy and disease monitoring. The 2013 ESH/ESC classification focuses on the classification of arterial hypertension (AH) based on cut-off systolic and/or diastolic BP values (Table 1).
Table 1. Classification of arterial hypertension
|
Category
|
Systolic
|
And/or
|
Diastolic
|
|
Optimal |
< 120 |
And |
< 80 |
|
Normal |
120-129 |
And/or |
80-84 |
|
High normal |
130-139 |
And/or |
85-89 |
|
Grade 1 Hypertension |
140-149 |
And/or |
90-99 |
|
Grade 2 Hypertension |
160-179 |
And/or |
100-109 |
|
Grade 3 Hypertension |
≥180 |
And/or |
≥110 |
|
Isolated systolic hypertension |
≥140 |
And |
< 90 |
Although the classification based on cut-off BP values is relevant to hypertension, it is difficult to apply to HHF patients because of a continuous relationship between BP and its associated complications such as CVD and renal events, which makes the distinction between normotensive and hypertensive patients challenging. In addition, in the general population, cut-off systolic and diastolic BP values assume a unimodal distribution and only a small percentage of the hypertensive population has elevated BP alone with the majority exhibiting additional CVD risk factors [10]. Since 1994 to date, the ESH, ESC and the European Atherosclerosis Society (EAS) developed joint recommendations on preventing CAD in clinical practice and emphasized that the prevention of CVD should be based on the quantification of global CVD risk. When concomitantly present, BP and other CVD risk factors potentiate each other and thus focusing on only BP may under-determine the actual HF risk in hypertensive patients [10].
The SSC proposed classification specific to HHF should be based on three common cardiac abnormalities: (a) ventricular; (b) ischemic and/or (c) arrhythmic acronymized as VIA [6]. Abnormalities in LV structure and function adopted the three main stages of hypertensive heart disease: (a) response of ventricular myocardium begins with electrocardiography (ECG) or echocardiography defined LVH; (b) then diastolic dysfunction may appear at times evolving to systolic dysfunction; and finally (c) HF development with non-specific symptoms. Ischemia in hypertensive patients is frequently results from microvascular dysfunction. If atherosclerosis developed, epicardial CAD and eventually acute coronary syndrome could be present. Finally, atrial fibrillation (AF) is the most common arrhythmia in hypertensive patients. Summarily, high BP patients could be classified based on the status and severity of ventricular dysfunction, ischemia and arrhythmias [6].
Epidemiology of HF has received extensive research but the etiology of HF in the contemporary population remains incompletely described. Epidemiological data on HHF is somewhat discordant and the prevalence of LVH in hypertensive patients varies significantly depending on the severity of hypertension – 20% in mild hypertension to 100% in severe or complicated hypertension [11]. In a post hoc analysis of the Framingham Heart Study (FHS) cohort, hypertension antedated the development of congestive heart failure (CHF) in 91% of the cases. Compared with normotensive patients, hypertension was associated with twice or thrice the risk of developing CHF after adjusting for age and other significant risk factors. It was also a significant risk factor for CHF in women (59%) and men (49%) but was associated with only 17% of hospitalized HF patients [12]. However, in an overview of 31 studies, hypertension was the primary etiological factor in only 4% of HF patients [13].
Two population-based studies implicate hypertension as the cause of 4% to 20% of CHF patients [14,15]. In a Swedish study recruiting 7,500 patients followed for 27 years, the main causes of CHF were hypertension (20%) and CAD either alone or in combination with hypertension (59%) [14]. In a South London study with a population of 292,000, incidences of HF were identified by prospectively monitoring patients admitted to a hospital through a rapid access HF clinic. Hypertension accounted for 4.4% of all incidents of CHF. Although, the study did not analyze the contribution of hypertension to HF patients with significant CAD, myocardial infarction (MI) was associated with a five-to-six fold increase in the risk of HF in hypertensive patients [16]. Antecedent hypertension interacted with neurohormonal activation and adversely altered early ventricular remodeling to elevate the risk of HF after MI [12].
In a more recent review of 30 echocardiographic studies involving 37,700 treated and untreated hypertensive patients, Cuspidi et al. [16] reported the prevalence rate of LVH in hypertensive patients was lower in untreated hypertensive cohorts (19% to 48%) and considerably higher in high-risk hypertensive patients (58% to 77%). The development of LVH is an early clinical manifestation of hypertension especially in children and adolescents [17]. Transient hypertension induced by mental stress or extensive increase in BP during exercise could also induce LVH. Similarly, the FHS also reported that LV mass can be increased prior to the development of overt hypertension [18]. The prevalence of LVH in hypertensive patients is not only influenced by an increase in LV afterload but also genetic components affecting hormonal activation pathways [19,20]. Other factors influenced influencing the prevalence of LVH in hypertensive patients include gender (with a higher proportion in women), obesity and possibly age [21]. Taken together, the current epidemiologic data on HHF demonstrates hypertension is the primary causative or exacerbating factor for HF but the absolute HF risk remains low in the absence of other significant risk factors [22].
Ventricular myocardial remodeling is an important clinical manifestation of HHF. The most important risk factor influencing myocardial remodeling is hypertension, and in the absence or presence of CAD, accounts for approximately 60% of HHF patients [14]. In addition to hypertension, factors such as ethnicity, gender, comorbidities such as obesity and diabetes, and environmental factors such as salt intake, and genetic factors may influence alterations in LV mass and geometry [23].
Obesity
The effect of obesity on cardiac structure is well established. Obesity induces an increase in LV mass (hypertrophy) independent of elevated BP levels and hypertension. Traditional explanations associates increased LV mass with obesity-induced hemodynamics alterations with a particular association with central fat distribution [24]. Higher central fat mass and the consequential increase in metabolic demand causes obese patients to exhibit elevated systemic blood volume, cardiac output and a redistribution of circulating blood volume to the cardiopulmonary region with a decrease in peripheral vascular resistance [23]. In addition, increasing circulating blood volume in obese patients could be a consequence of increased water retention due to high dietary salt intake associated with food overload. Increased circulating volume causes the LV to dilate suggesting an association between obesity and eccentric hypertrophy [25,26].
Non-hemodynamic factors involving inflammatory factors such as epicardial fat deposition, adipokines, lipotoxicity, sympathetic overdrive and activation of the renin–angiotensin–aldosterone system (RAAS) also contribute to obesity-associated LV remodeling. As a result, other structural alterations including fibrosis and increased epicardial fat deposition are frequent feature in the heart of obese patients leading to thicker LV walls and concentric geometry [26]. Obese patients with comorbid arterial hypertension often exhibit pressure overload, which has been associated with increased prevalence of LVH. The presence of pressure overload is clinically relevant since systemic hypertension and obesity are frequent comorbidities [23]. Several studies have reported increased LVH prevalence in obese patients. The prevalence ranges from 13% in normotensive obese patients to over 75% in hypertensive individual with morbid obesity [23]. In a cohort of 4,176 hypertensive patients, the prevalence of LVH was 12%, 25% and 48% in normal weight, overweight and morbid obesity respectively [26]. Findings of autopsy and prospective clinical trials also reveal a combination of concentric/eccentric LVH is common in hypertensive obese patients [27,28].
Diabetes
Data from experimental, pathologic, epidemiologic and clinical studies demonstrate diabetes mellitus (DM) increases the risk of HHF. Diabetes causes changes in cardiac structure and function independent of hypertension and CAD [29]. Interstitial fibrosis, cardiomyocyte hypertrophy and increased contractile protein glycosylation are common findings in biopsies of diabetic hearts [23]. Mechanisms of the changes in cardiac structure in diabetic hearts involve hyperglycemia, hyperinsulinemia, oxidative stress and RAAS activation. Compared to non-diabetic patients, diabetic individuals exhibit increased LV mass and wall thickness, even after adjusting for body mass index and BP [7,29]. However, the exact effect of diabetes on LV geometry remains incompletely understood because of the presence of concentric or eccentric remodeling in diabetic individuals [7]. Comorbid DM and systemic hypertension have an additive effect in increasing LV mass. In the Hypertension Genetic Epidemiology Network (HyperGEN) study recruiting a population-based cohort of 1950 hypertensive individuals, LVH was 32% higher in diabetic than in non-diabetic hypertensive individuals and the effect was independent of sex, BP and obesity [30].
Genetic
Hypertension, obesity and diabetes are important determinants of LVH but do not identify many individuals with the condition suggesting genetic factors may play a role in the development of LVH and consequently increase the risk of developing LVH [23]. Whilst much research has been undertaken to understand the causes of LVH and medical treatment to regress LVH, much remain unknown on its genetic basis. It has been proposed that LVH is a complex genetic disease likely to represent the interaction of several genes with the environment. The heritability of LV mass measured as a quantitative trait ranged between 30% and 70% in different populations suggesting a familial component [31]. Genes encoding proteins as well as those encoding cell signal transduction, hormones, growth factors, calcium homeostasis, substrate metabolism and BP are potential candidates involved in the development of different forms of LVH [31]. Various reports including candidate gene-association and genome-wide association studies have investigated common genetic variant associated with cardiac structure and suggest multiple variants each with modest effect size maybe involved in the modulation of LV mass [7]. However, results of these studies are insufficient to translate into clinical practice [31].
Ethnicity
Ethnicity may be potentially associated with increased risk of developing HHF. It influences the epidemiology of LVH. The prevalence of LVH and concentric hypertrophy is higher in blacks than in whites [7,11]. However, the current evidence is insufficient to suggest the relationship of ethnicity and LVH represent an independent effect of BP as blacks exhibit a higher risk of hypertension and higher BP levels among hypertensive subjects [27].
Salt intake
Salt intake has also been linked to increased risk of developing LVH and HHF. High salt intake assessed with 24-hour sodium excretion was an independent determinant of LV mass and is related to LV mass even after correcting 24-hour blood pressure in hypertensive patients but not in normotensive individuals [23]. A reduction in dietary sodium causes a reduction in LVH. In addition, salt intake also has a causal relationship with BP. A reduction in salt intake significantly lowers BP in both hypertensive and normotensive individuals [32]. However, the mechanism underlying the association between salt intake and LV remodeling is not well understood but the some explanations. High dietary sodium may lead to cardiac hypertrophy by promoting increases in BP and intravascular volume or by imposing direct effects on myocardial cells [23]. Salt intake may be associated with myocardial fibrosis by stimulating aldosterone synthesis and increase AT1 receptors in the myocardium [27].
Etiology
Myocardial remodeling secondary to chronic and progressive increase in blood pressure (BP) has been implicated as the primary etiology of HHF [4]. According to the 2007 ESC HF guidelines, objective evidence indicating diastolic dysfunction such as elevated diastolic filling pressure or decreased mitral annulus diastolic relaxation velocities, or reduced cardiac output support the clinical diagnosis of myocardial dysfunction [34]. HF patients with reduced ejection fraction (EF) typically exhibit progressive chamber dilatation, eccentric remodeling and systolic impairment because of reduced EF. Specific therapies targeting the reversal of structural and functional abnormalities reduce morbidity and mortality in these patients. However, the pathophysiology of HF with reduced EF in hypertensive patients remains partially understood [35]. In recent large clinical trials, the presence of concentric remodeling (or LVH), left atrial (LA) enlargement and diastolic dysfunction are the cardinal features of HF with reduced EF [36].
Concentric ventricular remodeling (or overt LVH) is a frequent occurrence in hypertensive individuals and is more likely associated with normal or even reduced LV end diastolic volume (LVEDV) accompanied by elevated ventricular stiffness and limited distensibility [37]. At the same time, eccentric hypertrophy has been defined by an increase in LV mass through larger LV diameter and LVEDV [38]. The development of LVH is a consequence of chronic BP and volume overload. To compensate for chronic BP in hypertensive subjects, LV wall thickness gradually increases to normalize wall stress resulting in concentric LV remodeling and LVH [37]. The activation of several biological processes including hormones pathways, growth factors and cytokines contribute to protein genesis by promoting growth of cardiomyocytes resulting structural alterations and remodeling [33]. Untreated hypertension may cause the progression of LVH to HF in the setting of serial events including ischemic, cardiomyocyte apoptosis or fibrosis and ultimately systolic dysfunction [39].
It was traditionally known that hypertension leads to concentric hypertrophy followed by chamber dilation and ultimately HF [23]. As early as 1982, it was reported that hypertrophic response of the failing heart involved a compensatory response followed by a progressive worsening of symptoms ending with the death of the patient due to degeneration and weakening of the myocardium. These pathologic mechanisms were reproduced in animal models of pressure overload due to aortic banding and in humans with aortic stenosis and hypertrophic cardiomyopathy [7]. Today, LVH has been shown to be an important intermediate phenotype in the progression of HHD to HHF [40]. Thus, the pathophysiology of HHF may be described in two phases: (a) the progression of hypertension to LVH; and (b) the progression of LVH to HF.
Hypertension to LVH
The development of concentric LVH is an important step in the pathway towards the development of HHF as illustrated in Figure 1.
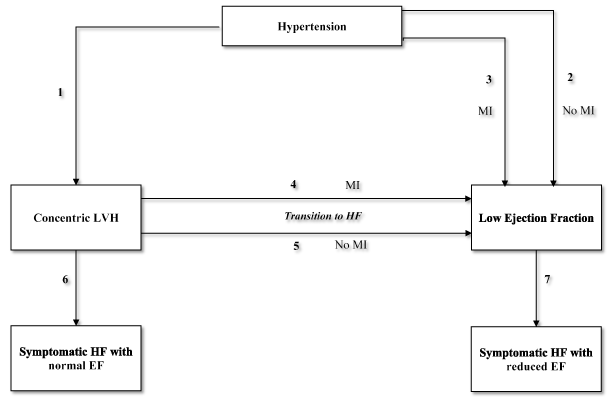
Figure 1. Pathways in the progression of hypertension to heart failure
The progression from hypertension to heart failure has seven pathways (1 to 7). The direct pathway from hypertension to HF with reduced ejection fraction may occur without pathway 2 or with pathway 3 (myocardial infarction [MI]). Usually, concentric ventricular remodeling transitions to HF through MI (pathway 4) and rarely without MI (pathway 5). Concentric LVH could develop symptomatic HF with reduced LVEF (pathway 7). Not shown in the diagram are other important risk factors for the development of HF include diabetes mellitus, age, environmental exposure and genetic factors. The thicker arrows indicated common pathways while the thinner arrows uncommon pathways. Reprodued from Drazner 2011, p. 328 [7]
The main pathological mechanisms contributing to the development of HHF are an increase in the size of cardiomyocyte size, alterations in the extracellular matrix (ECM) with accumulation of fibrosis and abnormalities of the intramyocardial coronary vasculature including medial hypertrophy and peri-vascular fibrosis [41,42]. While compensatory mechanisms in the setting of increased mechanical stress due to chronic high BP is the primary cause of progression from hypertension to LVH, other mechanisms such as neurohormones, growth factors and cytokines contribute to the progression to LVH [43]. Tighter control of systolic BP (target < 130 mmHg vs. 140 mmHg) is associated with a reduction in the development of LVH on ECG suggesting the importance of pressure load itself [44].
Factors other than chronic high BP may contribute to LV hypertrophic response since the increase in LV mass has considerable inter-individual variability. African Americans have a higher increase in LV mass and more severe diastolic dysfunction compared to whites [45-47]. Multi-variable models adjusted for known risk factors including systolic BP explain 50% of the variability in evaluated by echocardiography and 60% to 68% evaluated by cardiac magnetic resonance imaging [48]. These findings suggest other risk factors may contribute to the pathophysiology of HHF. In support, increasing evidence from epidemiological, sibling and observation studies suggest genetic factors modulate the development of HHF and may contribute to the inter-individual variability in hypertension-associated LVH [49,50].
Classification of lv hypertrophic response: The increase in LV mass (or structural changes in LV geometry) maybe classified into LV wall thickening due to pressure overload or chamber dilation due to volume overload [51]. The two classification patterns maybe categorized using relative wall thickness, defined as the ratio of LV wall thickness to diastolic diameter measured by echocardiography usually in response to volume overload. LV is classified as concentric when relative wall thickness is increased and eccentric when the relative wall thickness is not increased. Concentric remodeling occurs when relative wall thickness is increased but LV mass remains the same (Table 2) [52].
Table 2. LV classification based on lv mass and relative wall thickness
|
|
Normal Relative Wall Thickness |
Increased Relative Wall Thickness |
Normal LV mass |
Normal |
Concentric remodeling |
Increased LV mass |
Eccentric hypertrophy |
Concentric hypertrophy |
Echocardiographic studies demonstrate that hypertensive patients could have any of these three patterns LV geometry but it remains uncertain why some hypertensive patients develop concentric or eccentric hypertrophy [52,53]. The different LV-geometry patterns may be the result of the joint influences of volume and pressure overload, and contractile dysfunction [53-55]. Some of the variability of LV geometry in hypertensive patients may also be the consequence of differences to pressure overload itself including the severity, duration or rate of increase in BP [54,55]. Patients with concentric compared to eccentric hypertrophy exhibit a higher systolic BP and total peripheral resistance as well as higher ambulatory BP even when office BP is not significantly different [56-58].
Modulators of lv hypertrophic response: The main factors modulating LV hypertrophic response include demographic factors, comorbidities, neurohormonal activation and genetic factors [7,23]. Demographic factors (ethnicity, gender and age) modulate LV response to higher BP levels in hypertensive patients [7]. African Americans are more likely to develop concentric hypertrophy, women are more likely to develop concentric hypertrophy while men eccentric hypertrophy, and older age more likely to develop concentric hypertrophy [46,58,59]. Comorbidities may also contribute to variable LVH response to BP in hypertensive patients – CAD is associated with higher LV systolic dimension and eccentric hypertrophy [60]; DM with concentric hypertrophy [30]; and obesity with eccentric hypertrophy [60,61]. However, data on the association between comorbidities and LV hypertrophic response remain inconclusive [7].
Changes in neurohormonal activation in hypertensive patients may also contribute to the development of concentric or eccentric hypertrophy. High and low plasma renin activity common in hypertensive patients are associated with concentric and eccentric hypertrophy respectively [62].The Framingham Offspring Study investigating 2,119 patients associated increased aldosterone-to-renin ratio to both concentric and eccentric hypertrophy [63]. The findings suggest the importance of renin-angiotensin-aldosterone activity in predicting to development of increased LV wall thickness or ventricular dilation in hypertensive patients. Finally, changes in ECM may mediate in the development of LV chamber dilatation. In 39 hypertensive patients without CAD (16 with cardiac failure and 23 with preserved LV ejection fraction), endomyocardial biopsy of the RV septum showed cardiac failure patients had lower amount of collagen surrounding cardiomyocytes, higher amount of perivascular and scar-related collagen and a higher ratio of matrix metalloproteinase-1 (MMP1) to tissue inhibitor of MMP1 [64]. The findings suggest changes in ECM may contribute to LV dilation.
LVH to heart failure
In Figure 1, the classical paradigm suggests that in HHD patients, hypertension leads to concentric hypertrophy (Pathway 1), which then transitions into symptomatic HF with normal EF (pathway 6) or into dilated cardiac failure with reduced EF (pathway 7) in the setting of MI (pathway 4) or in the absence of MI (pathway 5) [7]. Current evidence links progressive alterations in ECM with the development of HF in patients with LVH and preserved EF [5,65]. In a canine model of hypertension, the administration of exogenous mineralocorticoid lead to a progressive cardiac fibrosis and increased LV stiffness suggesting its activation may contribute to this progression [66]. Changes in plasma levels of MMPs and tissue inhibitor of MMPs have also been linked with the progression of HHD [67,68]. Patients with LVH and cardiac failure exhibit higher MMPs tissue inhibitor linked to increased collagen deposition compared to hypertensive patients with no clinical HF [67]. High LV filling pressure may also contribute to the development of HF with preserved EF in hypertensive patients. Data from implantable hemodynamic monitors show patients with acute decompensated HF and chronic compensated HF have elevated LV filling pressure [69]. Echocardiographic data also demonstrate enlarged LA in HF patients with preserved EF and elevated pulmonary artery pressure, which correlate with LV filling pressure [70,71]. The present findings suggest that diastolic and systolic dysfunction are important mechanisms in the progression of LVH to HHF.
Diastolic dysfunction: Diastolic dysfunction is a major factor in the pathophysiology of HHD and its progression to symptomatic HHF [72]. About 40% of HHD patients have a normal systolic function (LVEF) but with an abnormal diastolic function [72,73]. Diastolic dysfunction is the main cause of the development of symptomatic HF in hypertensive patients [74]. LV diastolic dysfunction manifests as increased LV wall thickness and chronically elevated LV end-diastolic pressure leading to increased LA volume [5]. The increase in LV volume is the consequence of elevated LV filling pressure or LA pressure manifesting as exercise intolerance in HHF patients [72]. Myocardial ischemia is another key factor in the mechanism of leading to diastolic impairment in HHF while hypertension accelerates arteriosclerosis in both systemic and coronary arteries [75]. Chronic increase in LV wall stress and workload contribute to the development of LVH as well as is associated with an increase in the size of cardiomyocytes in the absence of a proportional proliferation of the capillary vasculature. These pathological changes result in ischemia (mismatch between myocardial oxygen demand and supply) in patients with longstanding hypertension [5,72]. The underlying myocardial ischemia and LVH mediates the association between HHF and relaxation abnormalities. HHF patients with impaired of LV pressure/volume reserve accompanied by impaired relaxation are usually asymptomatic during rest. However, a slight change in circulating volume or an increase in systemic vascular resistance during exertion makes their stiff LV incapable of handling increased circulating volume and ejecting appropriate cardiac volume leading to progressive decline in ventricular function and ultimately the development of HHF [72].
Systolic dysfunction: Systolic dysfunction, although not common, is a pathological mechanism observed in some HHF patients manifesting as HF with reduced EF. In the FHS, severe LV systolic dysfunction occurred in between 3% and 6% of patients with hypertension [76]. The Cardiovascular Health Study (CHS) [77] reported eccentric pattern of LVH is a significant risk factor for LV systolic dysfunction. Severe LV systolic dysfunction (LVEF < 30%) occurred on 6% of HF patients with hypertension. However, the study also reported that hypertension-induced LV remodeling or LVH was followed by chamber dilation and HF if not treated appropriately. Initially, LV dysfunction is compensatory but later followed by progressive worsening of symptoms ultimately ending with cardiac death [7]. The phenomenon of LVH followed by cardiac chamber dilation was reproduced in animal models of pressure overload in the setting of aortic banding and in humans with aortic stenosis and hypertrophic cardiomyopathy [5,7].
Left ventricular hypertrophy
Serial changes in LV mass (LVH) in HHF patients has been associated with poor prognosis [23]. Large prospective studies and meta-analysis have examined the prognostic impact of LVH in HHF patients. In the analysis of 20 prospective studies including 48,545 patients between 1960 and 2000 revealed the presence of LVH is associated with a 2.3 fold increase in the risk of CVD mortality and a 2.5 fold increase in the risk of CVD mortality [7]. In a post hoc analysis of 1,033 patients with uncomplicated hypertension from the Massa Ventricolare Sinistra nell’Ipertensione (MAVI) study, LVH was associated with increased risk of cardiovascular events (Relative Risk [RR] 2.80; 95% CI: 1.22-3.57). For every increase of 39 gm/m2 in LV mass, there is an independent 40% increase in the risk of major cardiovascular events in HHF patients [78]. In a meta-analysis of four studies including 1,064 hypertensive patients, the regression of LVH secondary to pharmacological control of BP improved cardiovascular outcomes and long-term prognosis. Compared to persistence or new development of LVH, regression of LVH was associated with 59% in the risk of cardiovascular events [79]. Several factors may mediate the prognostic impact of LVH in HHF patients. It has been suggested that hemodynamic and non-hemodynamic factors that promote increases in LV mass may also induce the progression and destabilization of atherosclerotic lesions. Thus, LVH may be considered a predictor for future cardiovascular events [23]. Alternatively, LVH may be a mediator of cardiovascular events by inducing MI and ischemia thereby predisposing HHF patients to arrhythmias and HF [80]. Already, LVH has been shown to antedate the development of HF in approximately 90% of HHF patients and increases the risk of HF by two-fold [27].
Left ventricular geometry
The geometry of the LV has also been shown to have a prognostic value in HHF patients. Patients with concentric hypertrophy have the highest risk of major cardiovascular events compared to patients with eccentric hypertrophy and concentric remodeling who have an intermediate risk between concentric and normal LV geometry [23]. Although the reasons why concentric hypertrophy has poor prognosis is not clear, it has been suggested that LV mass tends to be greater in patients with concentric compared to eccentric geometry. As a result, prognostic impact of concentric geometry could be the consequence of an overwhelming value of increased LV mass itself [80]. Patients with concentric geometry could also have been exposed to a more severe antecedent hypertension associated with increased rate of other end-organ damage and thus increased likelihood of cardiovascular events [23]. Increased relative wall thickness has an independent association with reduced flow reserve in hypertensive patients and may explain the increased risk of cardiovascular events in patients with concentric LV hypertrophy [81].
Patients with chronic systemic hypertension usually exhibit LVH, fibrosis, diastolic dysfunction and increased RAAS activation leading to congestive HF [82-84]. LV diastolic dysfunction, characterized by LV wall thickening and increased LA volume, is common in HHF patients leading to signs and symptoms including dyspnea, exercise intolerance, reduced quality of life, increased serum levels of natriuretic peptides and decreased functional and cognitive status [84]. Figure 2 illustrates the causes of LV diastolic dysfunction and its clinical consequences.
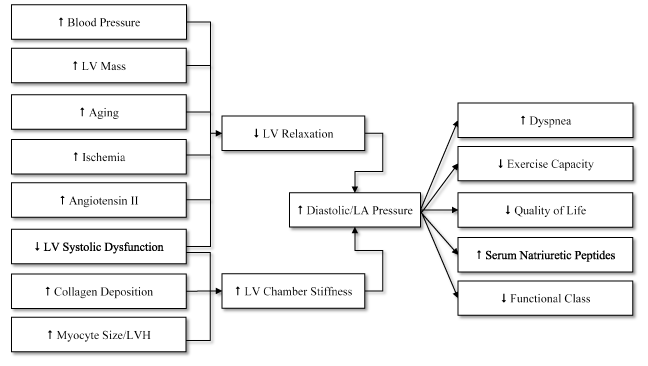
Figure 2. Causes of LV diastolic dysfunction and clinical consequences
Diastolic dysfunction may results from two pathologic conditions: (a) increased LV chamber stiffness due to increased collagen deposition and cardiomyocyte size/LVH; and (b) reduced LV relaxation due to increased BP, LV mass, aging, ischemic, angiotensin II, collagen and decreased LV systolic function. The clinical consequences of LV diastolic dysfunction include dyspnea, exercise intolerance, reduced quality of life, increased serum natriuretic peptides and decreased functional status. Reproduced from Phillips and Diamond, 2001, p. 487 [72]
In advanced stages, hypertensive patients may exhibit eccentric LVH and LV systolic dysfunction [84]. In support, post hoc analysis of Framingham Heart Study identifies LVH as a significant independent risk factor for cardiovascular morbidity and mortality [85]. HHF patients may also present with atrial fibrillation, whose incidence increases by between 40% and 50% in the presence of hypertension as well as increase frequency of ventricular arrhythmias [72].
Specific diagnostic guidelines for the assessment and detection of HHF are lacking. However, current evidence based on small-scale studies recommend pathological assessment remains an important procedure in the differential diagnosis of HHF [82]. Since LVH is the cardinal clinical manifestation of HHF and its identification is important for diagnostic and prognostic standpoint (identified hypertensive patients who may require more aggressive BP control), the basis of HHF diagnosis is detection of LVH assessed by ECG, endomyocardial biopsy and cardiac imaging using echocardiography and/or cardiac magnetic resonance imaging (CMRI).
Diagnostic methods
Electrocardiography: The 2013 ESH/ESC Guidelines for the management of arterial hypertension recommends that 12-lead electrocardiogram (ECG) in the initial or routine clinical evaluation of patients with hypertension to detect arrhythmias or evidence for ischemic heart disease and/or LVH [10]. ECG is useful in detecting patterns of ventricular overload or strain to suggest a more severe risk, ischemia, conduction abnormalities, LA dilatation and arrhythmias. 24-hour Holter ECG is indicated when arrhythmias and possible ischemic episodes are suspected [23]. The presence of S4 gallop suggests early diastolic dysfunction and possible LVH. Other abnormal ECG findings include LA enlargement, prolonged QT interval and LVH [82]. Although ECG parameters such as QRS duration and the Sokolow-Lyon index have been shown to independently predict cardiovascular events, ECG is valuable at least in patients > 55 years of age [10].
Endomyocardial biopsy: Diagnosis of HHF using invasive endomyocardial biopsy depends on the observation that HHF results from an increase in the quantity of myocardium as well as alteration in myocardial quality (fibrosis) [5]. Endomyocardial biopsy, although nowadays its utility is reducing, remains a powerful method for objective and specific assessment for LV abnormalities in HHF patients. In histopathological studies, LVH (cardiomyocyte hypertrophy) and moderate interstitial fibrosis were associated with HHF patients [86,87]. In HHF patients, LVH is a consequence of several pathological processes mediated by mechanical, neurohormonal and cytokines occurring in cardiomyocyte and non-cardiomyocyte compartments of the heart. Biopsy samples and postmortem of hearts of HHF patients reveal an exaggerated accumulation of fibers within the myocardial interstitium and surrounding intramural coronary arteries and arterioles [88,89].
Endomyocardial biopsy is useful in the quantification of diffuse myocardial fibrosis using collagen volume fraction. Compared to normotensive patients, the hearts of HHF patients exhibit significantly increased collagen volume fraction [86,88]. It has been proposed that myocardial fibrosis occurs by mechanical stress. Collagen volume fraction reflects transmural gradient of wall stress [90], the extent and severity parallels the increase in size of cardiomyocytes [91], and correlates with systolic blood pressure and pulse pressure in the myocardium of hypertensive patients [92]. Myocardial disarray defined as bundles of cardiomyocytes oriented perpendicularly or obliquely to each other or interspersed in different directions) common in patients with hypertrophic cardiomyopathy also appears in HHF patients but with a smaller distribution of myocardial disarray [93].
Echocardiography: Echocardiography is widely available and relatively low-cost non-invasive imaging method for the assessment of the morphology of LVH pattern in hypertensive patients [5]. Although both ECG and echocardiography can detect LVH, the sensitivity of ECG for LVH diagnosis is lower. In a review of ECG criteria for diagnosis of LVH in 4684 patients with persistent hypertension, Levy et al. [94] reported echocardiography detected LVH in 14.2% men and 17.6% women compared to ECG 2.9% men and 1.5% women. Echocardiographic LVH has a reported prevalence of 40% in hypertensive patients [95]. Echocardiographic assessment of LVH in hypertensive patients relies on evaluating LV mass, LV mass index and relative wall thickness. Using LV mass and relative wall thickness, LV geometry has been classified into four groups. (a) Concentric LVH: increased LV mass and increased relative wall thickness; (b) Eccentric LVH: increased LV mass but normal relative wall thickness; (c) Concentric remodeling: normal LV mass and increased relative wall thickness; and (d) Normal geometry: normal LV mass and normal relative wall thickness [95,96]. The American Society of Echocardiography (ASE) with the European Association of Echocardiography (EAE) [97] propose criteria for the assessment of LVH in hypertensive patients based on revised Simpson rule (Table 3).
Table 3. Reference limits and partition values of LV mass indexed to BSA (LV mass/BSA, g/m2)
|
Women |
Men |
Method |
Ref. Range |
Mildly Abnormal |
Moderately Abnormal |
Severely Abnormal |
Ref. Range |
Mildly Abnormal |
Moderately Abnormal |
Severely Abnormal |
|
Linear Method |
43-95 |
96-108 |
109-121 |
≥ 122 |
49-115 |
116-131 |
132-148 |
≥ 149 |
|
2D Method |
44-88 |
89-100 |
101-112 |
≥ 113 |
50-102 |
103-116 |
117-130 |
≥ 131 |
BSA: Body Surface Area; LV: Left Ventricular
In addition to morphological changes (LVH), echocardiography is also useful for the assessment of diastolic dysfunction, which is present in approximately 50% of hypertensive patients. It is important to assess and monitor changes in conventional Doppler echocardiography parameters such as peak early filling velocity (E), late diastolic filling velocity (A) and their ratio (E/A), and deceleration time. Patients with chronic hypertension and advanced HHF exhibit a pseudo-normalization of E/A ratio also referred to as restrictive physiology [5]. Tissue Doppler Imaging (TDI) is also useful for objective quantification of the LV function and early diastolic mitral annular velocity (E’) and the ration (E/E’) to assess the severity of diastolic dysfunction in HHF patients [5]. In evaluating both invasive and non-invasive assessment of diastolic dysfunction Kasner et al. [98] reported LV filling index of E/E’ latera; is the best for detecting diastolic dysfunction in HF patients with normal EF. TDI-defined strain and strain rate parameters and speckle tracking echocardiography have also been reported as useful modalities for detecting diastolic dysfunction. They are important in assisting in discriminating patients with HHF from other causes of LVH [93,99]. Since abnormalities in strain rate parameters may occur in subclinical diastolic dysfunction in hypertensive patients, TDI maybe a useful modality for disease prevention [100,101].
Cardiac magnetic resonance: Continued technical advances has firmly established cardiac magnetic resonance imaging (CMRI) in research and in cardiovascular medicine [82]. Whereas echocardiography is a faster, low-cost and more portable modality, CMRI provides superior reproducible image quality because unlike echocardiography it does not depend on symmetry on LV shape [102]. CMRI is able to quantify both LVH with high reproducibility and myocardial fibrosis with high spatial and contrast resolution. Takeda et al. [103] examined the power of CMRI in distinguishing cardiac amyloidosis, hypertrophic cardiomyopathy and HHD, all of which manifest with LVH and HF. CMRI provides 3D data on cardiac anatomy, function and tissue characterization, coronary and microvascular perfusion and valvular disease without using ionizing radiation. Myocardial fibrosis or infiltration could be assessed after the administration of gadolinium, which accumulates in regions of interstitial accumulation. The extent and pattern of LGE distinguishes between hypertrophic cardiomyopathy and HHF. The utility of CMRI in HHF diagnosis enables reproducible assessment of relative wall thickness and LV mass with greater accuracy, which is important in assessing small changes in LV mass over time important for assessing the benefit of pharmacologic treatment in HHF patients [82]. CMRI could also be an important therapeutic decision-making and prognostic tool since it represents an independent predictor of cardiac mortality [103,104].
Meta-analysis of prognostic value of echocardiography in HHF patients
CMRI is a useful modality for accurate assessment of LVH and myocardial fibrosis in HHF. Its superior reproducible image quality and ability to document small changes in LV mass makes the modality a useful prognostic marker for future cardiovascular events [102]. However, its low cost and limited availability has reduced its use. Echocardiography on the other is low-cost, portable and widely available modality used in the assessment of LVH and diastolic dysfunction in HHF patients [5]. While the diagnostic value of echocardiography has been demonstrated in hypertensive patients in a previous meta-analysis of 29 studies [105], its prognostic value remains understudied and partially understood. Thus, the present meta-analysis aims to examine the prognostic value of echocardiography based on the assessment of LVH changes in HHF patients.
Search criteria and inclusion: This systematic review and meta-analysis was performed according to published recommendations of the Preferred Reporting Items for Systematic Reviews and Meta-analyses Protocols (PRISMA-P) statement [106]. Search for pertinent studies was conducted on online database PubMed using the following key terms: hypertension, left ventricular hypertrophy, echocardiography, prognosis and cardiovascular risk or events. In addition, citations from list of articles meeting the inclusion criteria and review articles were searched to identify other relevant studies. Inclusion criteria for the selection of the final articles were studies that (a) published in peer-reviewed journals; (b) evaluated hypertensive patients (BP) > 140 mm Hg systolic or 90 mm Hg diastolic; (c) conducted echocardiographic assessment at baseline and during treatment; (d) provided data on cardiovascular events; and (e) compared patients with and without LVH. There was no restriction on the publication period or language. Two independent reviewers performed the literature search and any disagreement was resolved by consensus. The following variables were extracted: study characteristics (author, year), patient characteristics (number, mean age, and percentage of male patients), baseline and follow-up characteristics (BP, LVM and LVH) and outcomes (cardiovascular events, LVH prevalence and event rate) (Table 4).
Table 4. Studies characteristic on prognostic value of echocardiography-assessed LVH changes
|
Clinical Trials Included in the Meta-Analysis |
Study Characteristics |
Verdecchia et al. [107] |
Cipriano et al. [108] |
Koren et al. [109] |
Devereux et al. [110] |
Muiesan et al. [111] |
Pierdomenico et al. [112] |
Yasuno et al. [113] |
|
Year |
1998 |
2001 |
2002 |
2004 |
2007 |
2008 |
2009 |
|
Patients (N) |
430 |
311 |
172 |
941 |
436 |
387 |
955 |
|
Mean Age (yrs.) |
48 |
51 |
47 |
66 |
52 |
52 |
64 |
|
Male (%) |
54 |
64 |
63 |
59 |
57 |
60 |
59 |
|
Baseline Systolic BP |
153 |
167 |
152 |
174 |
157 |
158 |
142 |
|
Follow-up Systolic BP |
145 |
147 |
148 |
99 |
146 |
142 |
NR |
|
Baseline Diastolic BP |
98 |
102 |
94 |
98 |
98 |
99 |
NR |
|
Follow-up Diastolic BP |
91 |
89 |
92 |
82 |
91 |
88 |
NR |
|
Baseline LVM (g/BSA m2) |
109 |
130 |
107 |
123 |
26.5 |
57 |
142 |
|
Follow-up LVM (g/BSA m2) |
101 |
122 |
108 |
99 |
27 |
48.5 |
NR |
|
LV Definition g/m2 |
125 |
125 |
110/134 |
116/104 |
50/47 |
50/47 |
125 |
|
Echo FUP (in months) |
37 |
89 |
66 |
58 |
54 |
74 |
40 |
|
Decrease in LVH (n) |
285 |
52 |
91 |
443 |
209 |
242 |
155 |
|
Increase in LVH (n) |
145 |
109 |
81 |
NR |
227 |
145 |
418 |
|
CVD Events (n) Dec/Inc LVH |
15/16 |
28 |
11/23 |
104 |
21/61 |
15/44 |
20/67 |
|
Prevalence LVH (%) |
26 |
44 |
22 |
70 |
57 |
48 |
60 |
|
Event Rate: Decreased LVH |
1.58 |
1.1 |
8.8 |
NR |
0.97 |
1.06 |
NR |
|
Event Rate: Increased LVH |
6.27 |
1.87 |
19.8 |
NR |
1.87 |
4.4 |
NR |
|
LVH Regression (HR: 95% CI) |
0.18 (0.05- 0.68) p=0.004 |
NR |
NR |
0.78 (0.65-0.94) p=0.009 |
0.52 (0.28-0.96) p=0.039 |
0.62 (0.44-0.88) p=0.01 |
0.30 (0.13-0.71) p=0.006 |
BSA: Body Surface Area; CVD: cardiovascular; FUP: Follow-up; LV: Left Ventricular; LVM: Left Ventricular Mass; NR: Not Reported
Studies characteristics and outcomes: The initial online search yielded 187 studies but only seven studies [103-107] fulfilled the inclusion criteria. Table 4 shows the main characteristics of the studies included in this meta-analysis recruiting a combined population of 3,632 hypertensive patients with a slightly higher proportion of males (60%). The mean age of the patients at entry ranged between 48 [107] and 66 years [110] with mean age 55 years. The interval between baseline and follow-up echocardiographic LV assessment ranged between 37 [107] and 89 months [108] with a mean of 64 months. At baseline, the prevalence of LVH ranged between 20% [109] and 70% [110%] with a mean of 50%. The definition of LVH at baseline varied between studies. It was defined based on LV mass in grams (g) corrected by body surface area (BSA) in m2 – 125 g/m2 for both sex [107,108,113] and 110/134 g/m2 [109] and 116/104 gm2 [110] for male/female respectively. Two studies defined LVH as g/m2.7 50/47 g/m2.7 for male/female respectively [111,112]. At follow-up, most of the patients were receiving pharmacologic intervention (anti-hypertensive drugs) and/or lifestyle measures. Overall, there were 395 cardiovascular events. In Figure 3, LVH regression as defined by echocardiography is associated with decreased cardiovascular events compared to persistent or increased LVH (OR: 0.35, 95% CI: 0.203-0.626, p-0.003). The findings suggest the LVH regression assessed by echocardiography is a good marker of future cardiovascular events in HHF patients.
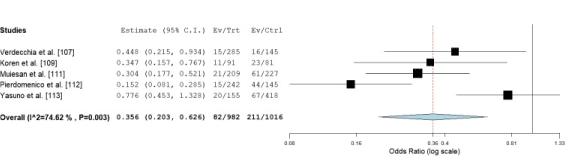
Figure 3. OR of Individual studies and pooled data on prognosis value LVH regression
Compared to persistent or increased LV hypertrophy, echocardiogrpahy defined LVH regression is predicts significantly lower cardiovascular events in future (OR: 0.35, 95% CI: 0.203-0.626, p-0.003)
Discussion: Echocardiographic-defined LVH (increase in LV mass) is a cardinal diagnostic feature of HF in hypertensive patients. In addition, serial changes in LV mass has also been described to have a prognostic value in HHF patients [7,23,77,78]. However, research evidence for the prognostic value of echocardiographic-defined serial changes in LV mass in HHF patients remain discordant and partially demonstrated. Since the use of echocardiography in the diagnosis of HHF is widespread, knowledge of its prognostic value would improve risk stratification of hypertensive individuals as well as guide the selection of appropriate treatment protocols and facilitate monitoring treatment efficacy [7,10]. The present meta-analysis sought to pool together pertinent studies on echocardiography-based assessment of LVH in hypertensive individuals to determine its prognostic value. This study finds serial changes in LV mass assessed by echocardiography could predict future cardiovascular events in hypertensive individuals. In particular, LVH regression (decrease in LV mass) was associated with 65% reduction in the risk of cardiovascular events compared to individuals with persistent or increased LVH in a long-term follow-up of 64 months.
The prognostic impact of LVH regression assessed by echocardiography in hypertensive individuals was initially demonstrated in an earlier meta-analysis of four studies including 1064 subjects. LVH regression reduced the risk of future cardiovascular by 59% relative to individuals with persistent or increased LVH but lacked multivariate analysis because of the characteristics of individual studies. Another recent meta-analysis including a larger sample (five studies with 3,149 hypertensive individuals) used adjusted statistics reported in individual studies to confirm and extend the findings on the prognostic value of LVH regression in hypertensive individuals assessed by echocardiography [114]. While LVH regression translates into better cardiovascular outcomes in HHF patients, the underlying mechanisms remain uncertain. There are reports associating LVH regression with reduced risk of cardiac event through improved coronary flow reserve, reduction in the incidence of arrhythmias and reversal of systolic and diastolic dysfunction [72,73].
The importance of the regression of LVH in HHF patients has been associated with favorable prognosis because it is a key pathophysiological factor of HHF. Increased LVH is a consequence of compensatory mechanisms of chronic and elevated BP, which contributes to diastolic failure. Thus, LVH regression improves diastolic function and relives its associated HF symptoms [72,73]. In addition, LVH is a marker for long-term exposure to BP and other risk factors in the atherogenic process. Thus, LVH regression may reduce the levels of activity of other risk factors and the progression of atherosclerosis to explain the decrease in cardiovascular events in HHF patients with regressed LVH [5]. While the present findings suggest the prognostic value of echocardiography in serial assessment of LVH, they should be used and interpreted with caution. Not all clinical data were available from individual studies and other sub-analyses could not be performed. Although the whole sample in this meta-analysis is sufficient for statistical analysis, samples used in individual studies varied significantly and some were relatively small and vulnerable to false positive or negative results. The follow-up period after the initiation of anti-hypertensive therapy varied greatly from 37 to 89 months, which could result in a bias. Finally, these revise focuses on HHF patients while most of the studies included in the review recruited hypertensive patients and did not distinguish between those with HHF and those without.
The 2013 ESH/ESC Guidelines for the management of arterial hypertension recommends treatment of HHF should include non-pharmacologic (lifestyle changes) and pharmacologic support targeting the regression of LVH [10]. Regressed LVH has been associated with improvement in diastolic function, coronary flow reserve and reduction in the risk of AF, CHF and cardiovascular mortality [82]. Thus, the primary goal of HHF therapy is to maintain arterial BP of 130/80 mm Hg or better, which is the cardinal pathologic factor precipitating or exacerbating the development of LVH in patients with chronic hypertension [10].
Clinical management approaches
Non-pharmacologic: The main non-pharmacologic strategies is lifestyle changes, whose target is to prevent hypertension. BP-lowering effects of targeted lifestyle changes could be as effective as drug monotherapy but its main drawback is low-level of adherence over time, which requires special action to overcome [115]. Appropriate lifestyle changes are both safe and efficacious in delaying or preventing hypertension in non-hypertensive individuals as well as contributes to BP reduction in hypertensive individuals who are already under medical therapy allowing reduction in the frequency or amount of anti-hypertensive dosage [116]. The 2013 ESH/ESC Guidelines recommend the following lifestyle measures (management of risk factors) to reduce BP in hypertensive patients: (a) salt restriction; (b) moderation of alcohol consumption; (c) high consumption of vegetables, fruits and low-fat diet; (d) weight reduction and maintenance; and (e) regular physical exercises. In a meta-analysis of 105 trials randomizing 6805 patients, Dickinson et al. [117] reported improved diet, aerobic exercise, alcohol and sodium restriction, and fish oil supplements significantly reduced systolic BP and improved diastolic function. In addition, smoking cessation is important to improve cardiovascular risk and because smoking imposes an acute pressor effect that may increase daytime ambulatory BP [10].
Pharmacologic support: The 2003 and 2007 ESH/ESC guidelines for clinical management of arterial hypertension reviewed a large number of randomized clinical trials of antihypertensive therapy and established the main therapeutic benefit is the lowering of BP, which is independent of the drugs used [118,119]. The updated 2013 ESH/ESC reconfirm that diuretics (thiazides, chlorthalidone and indapamide), beta-blockers, calcium antagonists, angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) are all suitable and efficacious for the initiation and maintenance of antihypertensive treatment as either monotherapy or combined therapy [10]. Whereas some meta-analyses claim superiority of one class of anti-hypertensive agents over another for some outcomes [120-122], the differences may be due to selection bias of trials since the largest available meta-analyses do not show clinically relevance differences between anti-hypertensive drug classes [123-125].
ACE-inhibitors and ARBs are particularly effective anti-hypertensive medication because angiotensin II has a demonstrated direct tropic effect on the pathogenesis of LVH [126,127]. A decrease in LV mass has been reported to occur in about three to six months following treatment initiation. The direct renin inhibitor aliskiren has also been found to facilitate the regression of LVH. However, the potential benefit of a dual therapy of ACE-inhibitor and ARBs with a direct renin effect is under investigation with initial trial reporting no evidence of an additive benefit on LVH regression [128]. ACE-inhibitors also appear to be beneficial to LVH patients because of improvement of in coronary endothelial function by bradykinin-mediated release of nitric oxide and decreased myocardial oxygen consumption through nitric oxide inhibition of mitochondrial respiration [129]. When initiating ACE-inhibitor therapy, it is important to monitor for angioedema, a life-threatening side effect, which occurs in a small number of patients. Patients who develop shortness of breath or swelling of the throat should discontinue ACE-inhibitor and admitted in emergence department since it is a potentially life-threatening side effect [82]. Patients with renal dysfunction such as renal artery stenosis require monitoring. If serum creatinine increases by more than 0.3 mg/dL from baseline, ACE-inhibitor should be discontinued and the reversible decrease will correct itself [82].
Calcium channel blockers facilitate LVH regression as well as improve coronary perfusion in LVH patients [130,121]. Calcium channel blockers that have a long-lasting action are safe and appropriate for treatment of hypertensive patients with LVH. Selection of calcium channel blockers should be guided by the patient’s resting heart rate and response to exercise. In patients with hyper-adrenergic response to exercise, recommended calcium channel blockers are verapamil or diltiazem, which provide symptomatic relief because they attenuate both heart rate and BP response to exercise. A dual therapy of ACE-inhibitor or ARBs with amlodipine (calcium channel blocker) may have an additive effect in regulating BP, facilitating LVH regressions and decreasing mortality [132].
The traditional beta-blockers only attenuate the heart response rate to exercise and could cause an accentuated systolic BP elevation during exercise due to unopposed alpha constriction [82]. However, carvedilol and labetalol that have alpha-blocking properties attenuate both heart rate and exercise response same to calcium channel blockers verapamil and diltiazem to provide symptomatic relief of dyspnea or angina as well as facilitate a slower heart rate allowing for better diastolic filling of the ventricle [10]. In older patients with a conduction system disease and slower heart rates, dihydropyridines that facilitate LVH regression such as amlodipine is recommended [132]. Current ESH/ESC treatment recommendations for hypertensive patients with LVH however are based on BP lowering effect and do not favor the selection of a particular beta-blocker over another except when avoiding beta-blockers with intrinsic sympathomimetic activity with the potential of increasing the risk of sudden death [10].
Diuretics remain the cornerstone of antihypertensive treatment and the first choice medication to start treatment in both the Joint National Committee (JNC) [133] and WHO/International Society of Hypertension Guidelines [134]. Low dose diuretics increase the anti-hypertensive effect of most anti-hypertensive medications. In older patients with stiffened vessels and volume sensitive hypertension, low-dose thiazide diuretics is effective but in patients with creatinine clearance < 50 mL/min it has less effective for volume control and antihypertensive effect [82]. In these patients, chlorthalidone or indapamide should be considered because they are effective despite reduced renal function. Loop diuretics can also be considered in patients with reduced renal function but to achieve anti-hypertensive effect usually requires to be given twice daily [82]. In patients with resistant hypertension, thiazide diuretic is recommended as part of anti-hypertensive therapy
Meta-analysis of clinical management methods
HF presents the final common pathway of the clinical history of several cardiovascular diseases as well as is a major healthcare problem globally [133,134]. The Framingham Heart Study (FHS) was the first large scale study with a long-term follow-up to implicate hypertension as a significant cause of HF independent of CAD [12]. Over the years, research into clinical management of HF has grown with a focus on preventing and managing predisposing conditions including hypertension. Antihypertensive medication has shown efficacy in preventing HF development and its incidence in hypertensive patients by lowering BP and improving diastolic and/or systolic function. However, the efficacy of various antihypertensive group of medications in preventing cardiovascular events in HHF patients remains inconclusive. Previous anti-hypertensive meta-analyses with limited comparisons between active treatments suggest the superiority of one group of antihypertensive medication over another [120-125,135] but the 2013 ESH/ESC guidelines report no group of anti-hypertensive medication is superior and associates the findings to biases such as selection bias and the lack of data on multivariate analysis in individual studies. The present meta-analysis seeks to compare the efficacy of antihypertensive medication on a composite of cardiovascular death, myocardial infarction, stroke or hospitalization in HHF patients.
Search criteria and inclusion: Online databases PubMed, Medline and EMBASE were searched using the following key words: hypertension, antihypertensive agents, placebo and cardiovascular risk. The limits of the search were prospective randomized controlled trials recruiting at least 200 subjects and published between 1996 and 2018. The selection of 1996 as the starting year for the search was arbitrary but influenced by the publication of a landmark analysis on the prevention of HF in hypertensive patients in the same year [136]. The inclusion criteria were studies that (a) recruited hypertensive patients; (b) randomized patients into treatment and control groups; (c) compared at least two antihypertensive medication groups or one group with placebo; and (d) reported quantifiable measures of outcomes of antihypertensive medication over time. The selection of the studies and extraction of data from the included studies was done by two investigators and there was no discrepancy observed. Mean and standard deviation was used to analyze categorical data. For continuous data, meta-analysis was performed on raw data to calculate OR and 95% CI. We used either fixed or random effect model based on the presence or absence of heterogeneity between individual studies. Statistical significance was defined as p < 0.05. Data extracted from the included studies were author, age of patients, antihypertensive medication given, number of patients per treatment group, outcomes based on number of patients affected and Hazard ratio and follow-up period (Table 5).
Table 5. Characteristics of included studies on antihypertensive medical therapy
1st Author [Ref #] |
Mean Age (yrs.) |
Female (%) |
Medication
(Study vs. Control) |
No. of Patients |
End-point |
Composite end-point (HR [95% CI]) |
p-value |
FUP (Months) |
Study |
Control |
Study |
Control |
Estacio et al. [137] |
57 |
33 |
CCB vs. ACE-I |
235 |
235 |
25 |
5 |
9.5 (2.3-21.4) |
0.001 |
60 |
UKPDS Group [138] |
56 |
46 |
ACE-I vs. BB |
358 |
400 |
75 |
59 |
1.14 (0.81-1.61) |
0.44 |
48 |
Hansson et al. [139] |
52 |
47 |
ACE-I vs. Diuretics/BB |
5,492 |
5,493 |
363 |
335 |
1.05 (0.90-1.22) |
0.52 |
73 |
Hansson et al. [140] |
76 |
33 |
BB/Diuretic Old vs. New |
2,213 |
4,401 |
221 |
438 |
0.99 (0.84-1.16) |
0.89 |
48 |
NICS-EH Study Group [141] |
67 |
33 |
Diuretic vs. CCB |
204 |
210 |
21 |
18 |
0.97 (0.51-1.84) |
0.932 |
60 |
ALLHAT Group [142] |
67 |
47 |
α-Blocker vs. Diuretic |
9,067 |
15,268 |
365 |
608 |
1.03 (0.90-1.17) |
0.71 |
40 |
Brown et al. [143] |
65 |
54 |
CCB vs. Diuretic |
3,157 |
3,164 |
200 |
182 |
1.10 (0.91-1.34) |
0.35 |
48 |
Hansson et al. [144] |
60 |
51 |
CCB vs. BB/Diuretic |
5,410 |
5,471 |
403 |
400 |
1.00 (0.87-1.15) |
0.97 |
54 |
Yusuf et al. [145] |
66 |
27 |
ACE-I vs. Placebo |
4,645 |
4,652 |
651 |
826 |
0.78 (0.70-0.86) |
0.001 |
60 |
Brenner et al. [146] |
60 |
38 |
ARBs vs. Placebo |
751 |
762 |
327 |
359 |
0.84 (0.02-0.28) |
0.02 |
54 |
Dahlof et al. [147] |
67 |
54 |
ARBs vs. BB |
4,605 |
4,588 |
508 |
588 |
0.87 (0.77-0.98) |
0.021 |
58 |
Black et al. [148] |
66 |
56 |
CCB vs. BB |
3,786 |
3,831 |
364 |
365 |
1.02 (0.88-1.18) |
0.77 |
36 |
Julius et al. [149] |
67 |
42 |
ARBs vs. CCB |
7,649 |
7,596 |
810 |
789 |
1.04 (0.94-1.15) |
0.49 |
50 |
Rahman et al. [150] |
63 |
47 |
CCB vs. Diuretic |
2,274 |
3,648 |
129 |
193 |
1.12 (0.89-1.40) |
NR |
59 |
Dahlof et al. [151] |
NR |
23 |
CCB vs. BB |
9,639 |
9,618 |
429 |
474 |
0.90 (0.79-1.02) |
0.1052 |
66 |
Suzuki et al. [152] |
NR |
56 |
ARBs vs. Placebo |
1,053 |
995 |
6 |
10 |
0.61 (0.41-0.84) |
0.05 |
37 |
Mochizuki et al. [153] |
65 |
34 |
ARBs vs. Placebo |
521 |
517 |
92 |
149 |
0.61 (0.47-0.79) |
0.0002 |
37 |
Beckett et al. [154] |
84 |
60 |
ACE-I vs. Placebo |
1,933 |
1,912 |
138 |
193 |
0.66 (0.53–0.82) |
0.001 |
22 |
Ontarget Investigators [155] |
66 |
27 |
ACE-I vs ARBs |
8,576 |
8,542 |
1,412 |
1,423 |
1.01 (0.94-1.09) |
0.004 |
56 |
Yusuf et al. [156] |
67 |
43 |
ARBs vs. Placebo |
2,954 |
2,972 |
465 |
504 |
0.87 (0.76-1.0) |
0.048 |
56 |
ACE-I: Angiotensin-Converting Enzyme-Inhibitor; ALLHAT: Antihypertensive And Lipid-Lowering Treatment to Prevent Heart Attack Trial ARBs: Angiotensin Receptor Blockers; Beta-blocker; CCB: Calcium Channel Blockers; FUP: Follow up Period; HR: Hazard Ratio; NICS-EH: National Intervention Cooperative Study in Elderly Hypertensives; UKPDS: UK Prospective Diabetes Study Group
studies characteristics and outcomes: Twenty studies (20) were meeting the inclusion criteria were included in this meta-analysis [137-156]. The pooled population from the 20 studies was 158,797 hypertensive patients randomized into treatment group (74,522) and control or placebo group (84,275). The patients were older (mean age = 65 years) with a slightly lower female proportion (43%). Patients were recruited if they were hypertensive (BP >150 mm Hg systolic and/or > 95 mm Hg diastolic) with one or more additional cardiovascular risk factor (higher BMI, cholesterol, smoking, diabetes and increased LV mass index). The main classes of antihypertensive medications investigated were calcium channel blockers, ACE-inhibitor, ARBs, and diuretics. Individual studies compared ACE-I/ARBs and placebo [145,146,152-154,156], calcium channel blockers with either ACE-I/ARBs [137,149] or diuretics [141,143,144,150] or beta-blocker [148], and ACE-I with beta-blocker [138,147]. Only one study compared conventional antihypertensive medication (atenolol, metoprolol, pindolol or hydrochlorothiazide plus amiloride) with newer medication (Enalapril, Lisinopril, Felodipine or isradipine) [109]. The mean follow-up period was 51 months (range 22 [154] to 73 [139]). The composite end-point was cardiovascular death, MI, stroke, HF-related hospitalization and renal dysfunction.
We performed a series of meta-analysis directly comparing antihypertensive drugs to placebo and different classes of anti-hypertensive drugs in HHF patients (Figure 4). The direct comparisons of six studies [145,146,152-154,156] comparing ACE-I/ARBs to placebo revealed ACE-I/ARBs reduce the risk of cardiovascular events in HHF patients by 24% (OR: 0.763, 95% CI: 0.653-0.875, p=0.012). Direct comparisons of different classes of antihypertensive medication revealed no significant differences in their ability to reduce the risk of cardiovascular events. ACE-I/ARBs vs. beta-blockers (OR: 0.96, 95% CI: 0.743-1.227); beta-blocker vs. diuretic (OR: 1.004, 95% CI: 0.847-1.190); calcium channel blockers vs. diuretics (OR: 1.06, 95% CI: 0.954-1.173); calcium channel blockers vs. beta-blockers (OR: 0.95, 95% CI: 0.845-1.061); ACE-I vs. ARBs (OR: 0,95, 95% CI: 0.910-1.069). In only one study [137] the ACE-I (Enalapril) had a superior effect than calcium channel blocker (nisoldipine) (OR: 5.5, 95% CI: 2.059-14.565) but the difference was not statistically significant. In addition, the study recruited patients with non-insulin dependent diabetes and hypertension and investigated the secondary clinical endpoint of fatal and non-fatal myocardial infarction. The study could also not distinguish whether the difference was due to the deleterious effect of nisoldipine or the protective effect of Enalapril or a combination of both.
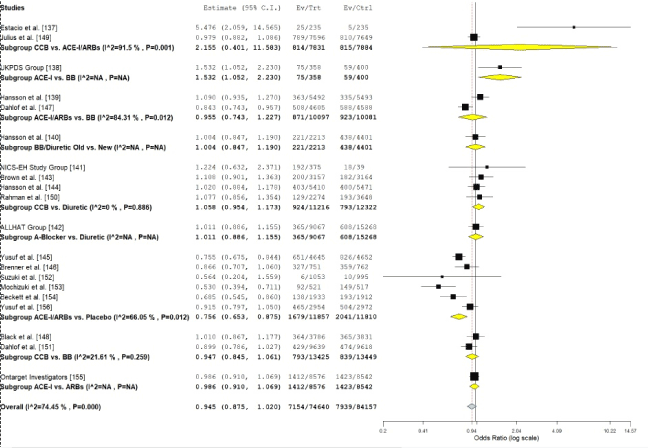
Figure 4. OR individual studies and pooled data on antihypertensive treatment
Sub group analysis (comparison) of odds ratio of individual studies grouped by type of anti-hypertensive medication
This meta-analysis demonstrates the use of any class of antihypertensive medication in HHF patients provides a protective effect against future cardiovascular events and the onset of HF compared to placebo. Further, this analysis based on combined studies on different hypertensive populations reveal that the different classes of hypertensive medication have comparable efficacy on the prevention of major cardiovascular events such as cardiovascular death, MI, stroke and HF-related hospitalization. Due to different studies using different sample sizes in the treatment and placebo groups, hypertensive patients (without demonstrated HHF), and the exclusion of the effect of differences in BP reduction in different therapies, it is essential to use and interpret these findings with caution. However, these findings reinforce current clinical guidelines for the treatment of HHF that recommend the use of anti-hypertensive medication on HHF patients despite other interventions such as lifestyle changes that may prevent or reduce the risk of developing HF [10,82]. The findings have an important clinical implication since hypertension is a major risk factor in the development of cardiovascular events particularly stroke in HHF patients and the incidence of stroke in these patients remain high despite antihypertensive treatment [157]. Thus, other risk factors or biological processes underlying the pathophysiology of cardiovascular events in HHF patients warrant further studies to improve treatment efficacy as well as to reduce their prevalence in HHF patients.
While the efficacy of antihypertensive medication is not disputed, in previous meta-analyses comparing individual hypertensive medications on the prevention of a single outcome suggest the superiority of one class over another. Chen et al. [158] analyzed 31 RCTs with 273,543 participants to investigate the efficacy of calcium channel blockers in the prevention of stroke in hypertensive patients. The study reported superiority of calcium channel blockers in reducing the incidence of stroke by 32% compared to placebo; 11% compared to a dual therapy of beta-blockers and diuretics; and 21% compared to beta-blockers alone. In a related network meta-analysis on the efficacy of antihypertensive medication in hypertensive patients, Sciarretta et al. [159] report diuretics, ACE-I and ARBs are the most effective first-line classes of antihypertensive medication to prevent the onset of HF compared to placebo and to calcium channel blockers together with beta-blockers and alpha-blockers. These findings suggest that different classes of antihypertensive medication may have better therapeutic effect on preventing some clinical outcomes such as stroke, onset of HF and CHD but not on composite end-points (outcomes). Despite the differences, the 2013 ESH/ESC guidelines on arterial hypertension recommend all the classes of antihypertensive medications for HHF patients because they have comparable efficacy in lowering BP and improving diastolic function, which are the cardinal factors underlying the pathophysiology and symptomatology of HHF respectively [10].
Hypertensive heart failure (HHF) is a cardiac condition characterized by myocardial abnormality resulting from long-standing arterial hypertension in the absence of any other cardiac disease capable of causing LVH or cardiac dysfunction. Its most common risk factors include obesity, diabetes mellitus, genetic predisposition, ethnicity (African-American) and high salt-intake. Its pathophysiologic mechanisms has been described based on the transition from hypertension to LVH and from LVH to HF through two main pathways. First, hypertension causing concentric LVH leading directly to symptomatic HF with normal EF or leading to symptomatic HF with reduced EF in the presence or absence of MI. Second, hypertension leading directly to depress EF with or without MI and ultimately symptomatic HF with reduced EF. The presence of LVH and concentric LV geometry are strong predictors of poor prognosis because of increased risk of major cardiovascular events. Its clinical manifestations include the typical signs and symptoms of HF such as dyspnea, exercise intolerance, reduced quality of life, increased serum levels of natriuretic peptides and decreased functional and cognitive status. Patients with chronic systemic hypertension may also present with LVH, fibrosis, diastolic dysfunction (LV wall thickening) and increased RAAS activation. Recommended diagnostic approach include ECG and cardiac imaging using echocardiography and cardiac magnetic resonance to assess for LVH and/or diastolic dysfunction. Endomyocardial biopsy is also useful in assessing the quantity of myocardium and alterations in myocardial quality (fibrosis) but its invasive nature limits its use. The target of clinical management is to decrease BP levels and relieve symptoms using anti-hypertensive medication (diuretics, beta-blockers, calcium antagonists, ACE-inhibitors and ARBs) and lifestyle changes (salt restriction, moderate alcohol consumption, weight reduction and regular physical exercises.
- Abraham WT (2013). Disease management: remote monitoring in heart failure patients with implantable defibrillators, resynchronization devices, and hemodynamic monitors. Europace 1: i40i4-6. [Crossref]
- LeMond L, Allen LA (2011). Palliative care and hospice in advanced heart failure. Prog. Cardiovasc. Dis 54:168-178. [Crossref]
- Moon JC, Treibel TA, Schelbert EB (2016). Myocardial fibrosis in hypertensive heart failure: does quality rather than quantity matter? J Am Coll Cardiol 67: 261-263. [Crossref]
- Ramakrishnan S, Kothari S, Bahl VK (2003). Hypertensive heart failure. Indian Heart J 55: 21-26 [Crossref]
- Kuroda K, Kato TS, Amano A. (2015). Hypertensive cardiomyopathy: A clinical approach and literature review. World J Hypertens 5: 41-52. [Crossref]
- Maqueda GI, Alegria-Ezquerra, E, Gonzalez-Juanatey JR. (2009). Hypertensive heart disease: a new clinical classification (VIA), E-Journal of the ESC Council for Cardiology Practice, 7. [Crossref]
- Drazner, M. H. (2011). The progression of hypertensive heart disease. Circ J 123: 327-334. [Crossref]
- Javier D. (2013). Hypertensive heart disease. Hypertens 152-166. [Crossref]
- Diamond JA, Phillips RA (2005). Hypertensive heart disease. Hypertens Res 28: 191-202. [Crossref]
- Council ES, Redon J, Narkiewicz K, Nilsson PM, Burnier M et al. (2013). 2013 ESH/ESC Guidelines for the management of arterial hypertension. Eur Heart J 34:2159-2219. [Crossref]
- Ruilope LM, Schmieder RE. (2008). Left ventricular hypertrophy and clinical outcomes in hypertensive patients. Am J Hypertens 21: 500-508. [Crossref]
- Levy D, Larson MG, Vasan R S, Kannel WB, Ho KK (1996). The progression from hypertension to congestive heart failure. Jama 275: 557-1562. [Crossref]
- Teerlink JR, Goldhaber SZ, Pfeffer MA (1991). An overview of contemporary etiologies of congestive heart failure. Am Heart J 121: 1852-1853. [Crossref]
- Wilhelmsen L, Rosengren A, Eriksson H, Lappas G (2001). Heart failure in the general population of men–morbidity, risk factors and prognosis. J. Intern. Med 249: 253-261. [Crossref]
- Fox KF, Cowie MR, Wood DA, Coats AJ, Gibbs JS, et al. (2001). Coronary artery disease as the cause of incident heart failure in the population. Eur Heart J 22: 228-236. [Crossref]
- Andersson B, Waagstein F. (1993). Spectrum and outcome of congestive heart failure in a hospitalized population. Am Heart J 126: 632-640. [Crossref]
- Cuspidi C, Sala C, Negri F, Mancia G, Morganti A (2012). Prevalence of left-ventricular hypertrophy in hypertension: An updated review of echocardiographic studies. J Hum Hypertens 26: 343-349. [Crossref]
- Post WS, Larson MG, Levy D (1994). Impact of left ventricular structure on the incidence of hypertension. The Framingham Heart Study. Circ 90: 179-185. [Crossref]
- Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ et al. (1998). Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science 280: 574-577. [Crossref]
- Schunkert H, Hense HW, Holmer SR, Stender M, Perz S et al. (1994). Association between a deletion polymorphism of the angiotensin-converting-enzyme gene and left ventricular hypertrophy. N Engl J Med 330: 1634-1638. [Crossref]
- Devereux RB, Roman MJ (1999). Left ventricular hypertrophy in hypertension: stimuli, patterns, and consequences. Hypertens Res 22:1-9. [Crossref]
- Richards AM, Nicholls MG, Troughton RW, Lainchbury JG, Elliott J et al. (2002). Antecedent hypertension and heart failure after myocardial infarction. J Am Coll Cardiol 39:1182-1188. [Crossref]
- Nadruz W (2015). Myocardial remodeling in hypertension. J Hum Hypertens 29: 1-6. [Crossref]
- Bays H. (2014). Central obesity as a clinical marker of adiposopathy; increased visceral adiposity as a surrogate marker for global fat dysfunction. Curr Opin Endocrinol Diabetes Obes 21: 345-351. [Crossref]
- Reisin E, Owen J (2015). Treatment: special conditions: Metabolic syndrome: obesity and the hypertension connection. J Am Soc Hypertens 9: 156-159. [Crossref]
- De Simone G, Izzo R, De Luca N, Gerdts E (2013). Left ventricular geometry in obesity: is it what we expect? Nutr Metab Cardiovasc Dis 23: 905-912.
- Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA et al. (2003). Seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertens 42: 1206-1252. [Crossref]
- Reisin E, Jack AV. Obesity and hypertension: mechanisms, cardio-renal consequences, and therapeutic approaches. Med. Clin North Am 93: 733-751. [Crossref]
- Murarka S, Movahed MR (2010). Diabetic cardiomyopathy. J Card Fail 16: 971-979. [Crossref]
- Palmieri V, Bella JN, Arnett DK, Liu JE, Oberman A et al. (2001). Effect of type 2 diabetes mellitus on left ventricular geometry and systolic function in hypertensive subjects: Hypertension Genetic Epidemiology Network (HyperGEN) study. Circ 103: 102-107. [Crossref]
- Arnett DK, de las Fuentes L, Broeckel U (2004). Genes for left ventricular hypertrophy. Curr Hypertens Rep 6: 36-41. [Crossref]
- He FJ, Burnier M, MacGregor GA (2011). Nutrition in cardiovascular disease: salt in hypertension and heart failure. Eur Heart J 32: 3073-3080.
- Lai YH, Lo CI, Wu YJ, Hung CL, Yeh HI (2013). Cardiac remodeling, adaptations and associated myocardial mechanics in hypertensive heart diseases. Acta Cardiol Sin 29: 64-70. [Crossref]
- Paulus WJ, Tschöpe C, Sanderson JE, Rusconi C, Flachskampf FA (2007). How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J 28: 2539-2550. [Crossref]
- Paulus WJ, van Ballegoij JJ (2010). Treatment of heart failure with normal ejection fraction: an inconvenient truth! J Am Coll Cardiol 55: 526-537. [Crossref]
- Zile MR, Gottdiener JS, Hetzel SJ, McMurray JJ, Komajda M (2011). Prevalence and significance of alterations in cardiac structure and function in patients with heart failure and a preserved ejection fraction. Circ 124: 2491-2501. [Crossref]
- Mayet J, Hughes A (2003). Cardiac and vascular pathophysiology in hypertension. Heart 89: 1104-1109. [Crossref]
- Verma A, Meris A, Skali H, Ghali JK, Arnold JM et al. (2008). Bourgoun M, Velazquez EJ, McMurray JJ, Kober L, Pfeffer MA, Califf RM. Prognostic implications of left ventricular mass and geometry following myocardial infarction: the VALIANT (VALsartan In Acute myocardial iNfarcTion) Echocardiographic Study. JACC Cardiovasc Imaging 1: 582-591. [Crossref]
- Fortuno MA, Ravassa S, Fortuño A, Zalba G, Díez J (2001). Cardiomyocyte apoptotic cell death in arterial hypertension: mechanisms and potential management. Hypertens 38: 1406-1412. [Crossref]
- Miura K, Dyer AR, Greenland P, Daviglus ML, Hill M, Liu K, Garside DB, Stamler J. Pulse pressure compared with other blood pressure indexes in the prediction of 25-year cardiovascular and all-cause mortality rates: The Chicago Heart Association Detection Project in Industry Study. Hypertens 38: 232-237. [Crossref]
- Berk BC, Fujiwara K, Lehoux S (2007). ECM remodeling in hypertensive heart disease. J Clin Invest 117: 568-575. [Crossref]
- Spinale FG (2007). Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 87: 1285-1342. [Crossref]
- Hill JA, Olson EN (2008). Cardiac plasticity. N Engl J Med 358: 1370-1380. [Crossref]
- Verdecchia P, Staessen JA, Angeli F, De Simone G, Achilli A, et al. (2009) Usual versus tight control of systolic blood pressure in non-diabetic patients with hypertension (Cardio-Sis): an open-label randomised trial. The Lancet 374: 525-533. [Crossref]
- Kizer JR, Arnett DK, Bella JN, Paranicas M, Rao DC et al. (2004). Differences in left ventricular structure between black and white hypertensive adults: the Hypertension Genetic Epidemiology Network study. Hypertens 43: 1182-1188. [Crossref]
- Drazner MH, Dries DL, Peshock RM, Cooper RS, Klassen C (2005). Left ventricular hypertrophy is more prevalent in blacks than whites in the general population: the Dallas Heart Study. Hypertens 46: 124-129. [Crossref]
- Sharp A, Tapp R, Francis DP, Thom SA, Hughes AD (2008). Stanton AV, Zambanini A, Chaturvedi N, Byrd S, Poulter NR, Sever PS. Ethnicity and left ventricular diastolic function in hypertension: an ASCOT (Anglo-Scandinavian Cardiac Outcomes Trial) substudy. J Am Coll Cardiol 52: 1015-1021. [Crossref]
- Devereux RB, Roman MJ, De Simone G, O’Grady MJ, Paranicas M et al. (1997). Relations of left ventricular mass to demographic and hemodynamic variables in American Indians: the Strong Heart Study. Circ 96: 1416-1423. [Crossref]
- Heckbert SR, Post W, Pearson GD, Arnett DK, Gomes AS (2006). Traditional cardiovascular risk factors in relation to left ventricular mass, volume, and systolic function by cardiac magnetic resonance imaging: the Multiethnic Study of Atherosclerosis. J Am Coll Cardiol 48: 2285-2292. [Crossref]
- Post WS, Larson MG, Myers RH, Galderisi M, Levy D (1997). Heritability of left ventricular mass: the Framingham Heart Study. Hypertens 30: 1025-1028. [Crossref]
- Frohlich ED, Apstein C, Chobanian AV, Devereux RB, Dustan HP (1992). The heart in hypertension. N Engl J Med 327: 998-1008. [Crossref]
- Sehgal S, Drazner MH (2007). Left ventricular geometry: does shape matter? Am Heart J 153: 153-155. [Crossref]
- Ganau A, Devereux RB, Roman MJ, De Simone G, Pickering TG (1992). Patterns of left ventricular hypertrophy and geometric remodeling in essential hypertension. J Am Coll Cardiol 19: 1550-1558. [Crossref]
- Ganau A, Devereux RB, Pickering TG, Roman MJ, Schnall PL et al (1990). Relation of left ventricular hemodynamic load and contractile performance to left ventricular mass in hypertension. Circ 81: 25-36. [Crossref]
- Devereux RB, Ganau A, Roman MJ (1994). Left ventricular hypertrophy and geometric remodeling in hypertension: stimuli, functional consequences and prognostic implications. J Hypertens Suppl 12: S117-S127. [Crossref]
- Ross, Jr J (1997). On variations in the cardiac hypertrophic response to pressure overload. Circ 95: 1349-1351. [Crossref]
- Fagard RH, Staessen JA, Thijs L (1997). Prediction of cardiac structure and function by repeated clinic and ambulatory blood pressure. Hypertens 29: 22-29. [Crossref]
- Devereux RB, James GD, Pickering TG (1993). What is normal blood pressure? Comparison of ambulatory pressure level and variability in patients with normal or abnormal left ventricular geometry. Am J Hypertens 6: 211S-215S. [Crossref]
- Chahal NS, Lim TK, Jain P, Chambers JC, Kooner JS, Senior R (2009). New insights into the relationship of left ventricular geometry and left ventricular mass with cardiac function: a population study of hypertensive subjects. Eur Heart J 31: 588-594. [Crossref]
- Zabalgoitia M, Berning J, Koren MJ, Støylen A, Nieminen MS (2001). Impact of coronary artery disease on left ventricular systolic function and geometry in hypertensive patients with left ventricular hypertrophy (the LIFE study). Am J Cardiol 88: 646-650. [Crossref]
- Gottdiener JS, Reda DJ, Materson BJ, Massie BM, Notargiacomo A (1994). Importance of obesity, race and age to the cardiac structural and functional effects of hypertension. J Am Coll Cardiol 24: 1492-1498. [Crossref]
- Du Cailar G, Pasquie JL, Ribstein J, Mimran A (2000). Left ventricular adaptation to hypertension and plasma renin activity. J Hum Hypertens 14: 181-188. [Crossref]
- Velagaleti RS, Gona P, Levy D, Aragam J, Larson MG et al. (2008). Relations of biomarkers representing distinct biological pathways to left ventricular geometry. Circ 118: 2252-2258. [Crossref]
- López B, González A, Querejeta R, Larman M, Díez J et al. (2006). Alterations in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure. J Am Coll Cardiol 48: 89-96. [Crossref]
- Goh VJ, Le TT, Bryant J, Wong JI, Su B et al. (2017). Novel index of maladaptive myocardial remodeling in hypertension. Circ Cardiovasc Imaging 10:e006840. [Crossref]
- Shapiro BP, Owan TE, Mohammed S, Kruger M, Linke WA et al. (2008). Burnett Jr JC, Redfield MM. Mineralocorticoid signaling in transition to heart failure with normal ejection fraction. Hypertens 51: 289-295. [Crossref]
- Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD et al. (2006). Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circ 113: 2089-2096. [Crossref]
- Essa EM, Zile MR, Stroud RE, Rice A, Gumina RJ et al. (2012). Changes in Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of MMPs Plasma Profiles in Stress-Induced Cardiomyopathy. J Card Fail 18: 487-492. [Crossref]
- Zile MR, Bennett TD, St John Sutton M, Cho YK, Adamson PB et al (2008). Transition from chronic compensated to acute decompensated heart failure: pathophysiological insights obtained from continuous monitoring of intracardiac pressures. Circ 118:1433-1441. [Crossref]
- Melenovsky V, Borlaug BA, Rosen B, Hay I, Ferruci L et al. (2007). Cardiovascular features of heart failure with preserved ejection fraction versus nonfailing hypertensive left ventricular hypertrophy in the urban Baltimore community: the role of atrial remodeling/dysfunction. J Am Coll Cardiol 49: 198-207. [Crossref]
- Lam CS, Roger VL, Rodeheffer RJ, Borlaug BA, Enders FT et al. (2009). Pulmonary hypertension in heart failure with preserved ejection fraction: a community-based study. J Am Coll Cardiol 53: 1119-1126. [Crossref]
- Phillips RA, Diamond JA (2001). Diastolic function in hypertension. Curr Cardiol Rep 3: 485-497. [Crossref]
- Aurigemma GP, Gottdiener JS, Shemanski L, Gardin J, Kitzman D (2001). Predictive value of systolic and diastolic function for incident congestive heart failure in the elderly: the cardiovascular health study. J Am Coll Cardiol 37: 1042-1048. [Crossref]
- Gandhi SK, Powers JC, Nomeir AM, Fowle K, Kitzman DW, Rankin KM, Little WC (2001). The pathogenesis of acute pulmonary edema associated with hypertension. N Engl J Med 344: 17-22. [Crossref]
- Chobanian AV, Brecher PI, Haudenschild CC (1986). Effects of Hypertension and of Antihypertensive Therapy on Atherosclerosis State of the Art Lecture. Hypertens 8: I-15. [Crossref]
- Devereux RB, Bella JN, Palmieri V, Oberman A, Kitzman DW et al. (2001). Hopkins PN, Rao DC, Morgan D, Paranicas M, Fishman D, Arnett DK. Left ventricular systolic dysfunction in a biracial sample of hypertensive adults: the HyperGen study. Hypertens 38: 417-423. [Crossref]
- Wang TJ, Evans JC, Benjamin EJ, Levy D, LeRoy EC et al. (2003). Natural history of asymptomatic left ventricular systolic dysfunction in the community. Circ 108: 977-982. [Crossref]
- Verdecchia P, Carini G, Circo A, Dovellini E, Giovannini E, et al. (2001) Left ventricular mass and cardiovascular morbidity in essential hypertension: the MAVI study. J Am Coll Cardiol 38: 1829-1835. [Crossref]
- Verdecchia P, Angeli F, Borgioni C, Gattobigio R, De Simone G, et al. (2003) Changes in cardiovascular risk by reduction of left ventricular mass in hypertension: a meta-analysis. Am J Hypertens 16: 895-899. [Crossref]
- Verdecchia P, Angeli F, Achilli P, Castellani C, Broccatelli A, et al. (2007) Echocardiographic left ventricular hypertrophy in hypertension: marker for future events or mediator of events? Curr Opin Cardiol 22: 329-334. [Crossref]
- Schafer S, Kelm M, Mingers S, Strauer BE (2002) Left ventricular remodeling impairs coronary flow reserve in hypertensive patients. J hypertens 20: 1431-1437. [Crossref]
- Katholi RE, Couri DM (2011). Left ventricular hypertrophy: major risk factor in patients with hypertension: update and practical clinical applications. Int J Hypertens 2011: 495349 [Crossref]
- Gonzalez A, López B, Dı́ez J (2004). Fibrosis in hypertensive heart disease: role of the renin-angiotensin-aldosterone system. Medical Clinics 88: 83-97. [Crossref]
- Gardin JM, Lauer MS (2004). Left ventricular hypertrophy: the next treatable, silent killer? Jama 292: 2396-2398. [Crossref]
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP (1990). Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 322: 1561-1566. [Crossref]
- Díez J, González A, López B, Querejeta R (2005). Mechanisms of disease: pathologic structural remodeling is more than adaptive hypertrophy in hypertensive heart disease. Nature Reviews Cardiology 2: 209-216. [Crossref]
- Díez J. (2008). Diagnosis and treatment of myocardial fibrosis in hypertensive heart disease. Circ 72: A8-12. [Crossref]
- Díez J (2007). Mechanisms of cardiac fibrosis in hypertension. J Clin Hypertens 9: 546-550. [Cossref]
- Brilla CG, Funck RC, Rupp H (2000). Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circ 102: 1388-1393. [Crossref].
- Tanaka M, Fujiwara H, Onodera T, Wu DJ, Hamashima Y et al. (1986). Quantitative analysis of myocardial fibrosis in normal, hypertensive hearts, and hypertrophic cardiomyopathy. Heart 55: 575-581. [Crossref]
- Rossi MA (1998). Pathologic fibrosis and connective tissue matrix in left ventricular hypertrophy due to chronic arterial hypertension in humans. J Hypertens 16: 1031-1041. [Crossref]
- Querejeta R, Varo N, López B, Larman M, Artinano E et al. (2000). Serum carboxy-terminal propeptide of procollagen type I is a marker of myocardial fibrosis in hypertensive heart disease. Circ 101: 1729-1735. [Crossref]
- Kato TS, Noda A, Izawa H, Yamada A, Obata K et al. (2004). Nagata K, Iwase M, Murohara T, Yokota M. Discrimination of nonobstructive hypertrophic cardiomyopathy from hypertensive left ventricular hypertrophy on the basis of strain rate imaging by tissue Doppler ultrasonography. Circ 110: 3808-3814. [Crossref]
- Levy D, Labib SB, Anderson KM, Christiansen JC, Kannel WB, et al. (1990) Determinants of sensitivity and specificity of electrocardiographic criteria for left ventricular hypertrophy. Circ 81: 815-820. [Crossref]
- Janardhanan R, Kramer CM (2011) Imaging in hypertensive heart disease. Expert Rev Cardiovasc Ther 9: 199-209. [Crossref]
- Foppa M, Duncan BB, Rohde LE (2005) Echocardiography-based left ventricular mass estimation. How should we define hypertrophy? Cardiovasc ultrasound 3: 17. [Crossref]
- Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, et al. (2005) Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 18: 1440-1463. [Crossref]
- Kasner M, Westermann D, Steendijk P, Gaub R, Wilkenshoff U, et al. (2007) Utility of Doppler echocardiography and Tissue Doppler imaging in the estimation of diastolic function in heart failure with normal ejection fraction: a comparative Doppler-conductance catheterization study. Circ 116: 637-647. [Crossref]
- Kato TS, Izawa H, Komamura K, Noda A, Asano H, et al. (2008) Heterogeneity of regional systolic function detected by tissue Doppler imaging is linked to impaired global left ventricular relaxation in hypertrophic cardiomyopathy. Heart. [Crossref]
- Narayanan A, Aurigemma GP, Chinali M, Hill JC, Meyer TE, et al. (2009) Cardiac mechanics in mild hypertensive heart disease: a speckle-strain imaging study. Circulation: Cardiovascular Imaging 2: 382-390. [Crossref]
- Solomon SD, Janardhanan R, Verma A, Bourgoun M, Daley WL, et al. (2007) Effect of angiotensin receptor blockade and antihypertensive drugs on diastolic function in patients with hypertension and diastolic dysfunction: a randomised trial. The Lancet 369: 2079-2087. [Crossref]
- Moon JC, Fisher NG, McKenna WJ, Pennell DJ (2004) Detection of apical hypertrophic cardiomyopathy by cardiovascular magnetic resonance in patients with non-diagnostic echocardiography. Heart 90: 645-649. [Crossref]
- Takeda M, Amano Y, Tachi M, Tani H, Mizuno K, et al. (2013) MRI differentiation of cardiomyopathy showing left ventricular hypertrophy and heart failure: differentiation between cardiac amyloidosis, hypertrophic cardiomyopathy, and hypertensive heart disease. Jpn J Radiol 31: 693-700. [Crossref]
- Martos R, Baugh J, Ledwidge M, O’Loughlin C, Conlon C, et al. (2007) Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circ 115: 888-895. [Crossref]
- Janda S, Shahidi N, Gin K, Swiston J (2011) Diagnostic accuracy of echocardiography for pulmonary hypertension: a systematic review and meta-analysis. Heart 1:hrt-2010. [Crossref]
- Moher D, Shamseer L, Clarke M, Ghersi D, Liberati A,et al.(2006) Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Systematic reviews 4: 1. [Crossref]
- Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, et al. (1998) Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circ 97: 48-54. [Crossref]
- Cipriano C, Gosse P, Bemurat L, Mas D, Lemetayer P, et al. (2001) Prognostic value of left ventricular mass and its evolution during treatment in the Bordeaux cohort of hypertensive patients. Am J Hypertens 14: 524-529. [Crossref]
- Koren MJ, Ulin RJ, Koren AT, Laragh JH, Devereux RB (2002) Left ventricular mass change during treatment and outcome in patients with essential hypertension. Am J Hypertens 15: 1021-1028. [Crossref]
- Devereux RB, Wachtell K, Gerdts E, Boman K, Nieminen MS, et al. (2004) Prognostic significance of left ventricular mass change during treatment of hypertension. Jama 292: 2350-2356. [Crossref]
- Muiesan ML, Salvetti M, Paini A, Monteduro C, Galbassini G, et al. (2007) Inappropriate left ventricular mass changes during treatment adversely affects cardiovascular prognosis in hypertensive patients. Hypertens 49: 1077-1083. [Crossref]
- Pierdomenico SD, Lapenna D, Cuccurullo F (2008) Regression of echocardiographic left ventricular hypertrophy after 2 years of therapy reduces cardiovascular risk in patients with essential hypertension. Am J Hypetens 21: 464-470. [Crossref]
- Yasuno S, Ueshima K, Oba K, Fujimoto A, Ogihara T, et al. (2009) Clinical significance of left ventricular hypertrophy and changes in left ventricular mass in high-risk hypertensive patients: a subanalysis of the Candesartan Antihypertensive Survival Evaluation in Japan trial. J hypertens 27: 1705-1712. [Crossref]
- Pierdomenico SD, Cuccurullo F (2010) Risk reduction after regression of echocardiographic left ventricular hypertrophy in hypertension: a meta-analysis. Am J Hypertens 23: 876-881. [Crossref]
- Elmer PJ, Obarzanek E, Vollmer WM, Simons-Morton D, Stevens VJ, et al. (2006) Effects of comprehensive lifestyle modification on diet, weight, physical fitness, and blood pressure control: 18-month results of a randomized trial. Annals of internal medicine 144: 485-495. [Crossref]
- Frisoli TM, Schmieder RE, Grodzicki T, Messerli FH (2011) Beyond salt: lifestyle modifications and blood pressure. Eur Heart J 32: 3081-3087. [Crossref]
- Dickinson HO, Mason JM, Nicolson DJ, Campbell F, Beyer FR, et al. (2006) Lifestyle interventions to reduce raised blood pressure: a systematic review of randomized controlled trials. J hypertens 24: 215-233. [Crossref]
- Guidelines Committee (2003). 2003 European Society of Hypertension–European Society of Cardiology guidelines for the management of arterial hypertension. J hypertens 21: 1011-1053. [Crossref]
- Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, et al. (2007) 2007 Guidelines for the management of arterial hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 28: 1462-536. [Crossref]
- Psaty BM, Lumley T, Furberg CD, Schellenbaum G, Pahor M, et al. (2003) Health outcomes associated with various antihypertensive therapies used as first-line agents: a network meta-analysis. Jama 289: 2534-2544. [Crossref]
- Costanzo P, Perrone-Filardi P, Petretta M, Marciano C, Vassallo E, et al. (2009) Calcium channel blockers and cardiovascular outcomes: a meta-analysis of 175 634 patients. J Hypertens 27: 1136-1151. [Crossref]
- Van Vark LC, Bertrand M, Akkerhuis KM, Brugts JJ, Fox K, et al. (2012) Angiotensin-converting enzyme inhibitors reduce mortality in hypertension: a meta-analysis of randomized clinical trials of renin–angiotensin–aldosterone system inhibitors involving 158 998 patients. Eur Heart J 33: 2088-2097. [Crossref]
- Law M, Morris JK, Wald NJ (2009) Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta-analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. Bmj 338: b1665. [Crossref]
- Trialists’Collaboration BP (2005) Effects of different blood pressure-lowering regimens on major cardiovascular events in individuals with and without diabetes mellitus: results of prospectively designed overviews of randomized trials. Arch Intern Med 165: 1410-1419. [Crossref]
- Blood Pressure Lowering Treatment Trialists' Collaboration (2003) Effects of different blood-pressure-lowering regimens on major cardiovascular events: results of prospectively-designed overviews of randomised trials. The Lancet 362: 1527-1535. [Crossref]
- Iriarte M, Caso R, Murga N, Boveda J, Saenz R, et al. (1995) Enalapril-induced regression of hypertensive left ventricular hypertrophy, regional ischemia, and microvascular angina. Am J Cardiol 75: 850-852. [Crossref]
- Mathew J, Sleight P, Lonn E, Johnstone D, Pogue J, et al. (2001) Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circ 104:1615-1621. [Crossref]
- Solomon SD, Appelbaum E, Manning WJ, Verma A, Berglund T, et al. (2009) Effect of the direct renin inhibitor aliskiren, the angiotensin receptor blocker losartan, or both on left ventricular mass in patients with hypertension and left ventricular hypertrophy. Circ 119: 530-537. [Crossref]
- Zhang X, Xie YW, Nasjletti A, Xu X, Wolin MS, et al. (1997) ACE inhibitors promote nitric oxide accumulation to modulate myocardial oxygen consumption. Circ 95: 176-182. [Crossref]
- Frishman WH, Skolnick AE, Strom JA (1989) Effects of calcium entry blockade on hypertension-induced left ventricular hypertrophy. Circ 80: IV151-61. [Crossref]
- Vogt M, Strauer BE (1995) Response off hypertensive left ventricular hypertrophy and coronary microvascular disease to calcium antagonists. Am J Cardiol 76: 24D-30D. [Crossref]
- Tapp RJ, Sharp A, Stanton AV, O'Brien E, Chaturvedi N, Poulter NR, Sever PS, Thom SA, Hughes AD, Mayet J, ASCOT Investigators. Differential effects of antihypertensive treatment on left ventricular diastolic function: an ASCOT (Anglo-Scandinavian Cardiac Outcomes Trial) substudy. J Am Coll Cardiol 55: 1875-1881. [Crossref]
- Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, et al. (2003) Seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertens 42: 1206-1252. [Crossref]
- World Health Organization, International Society of Hypertension Writing Group (2003) 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J hypertens 21: 1983-1992. [Crossref]
- Fagard RH, Cornelissen VA (2007) Incidence of cardiovascular events in white-coat, masked and sustained hypertension versus true normotension: a meta-analysis. J Hypertens 25: 2193–2198. [Crossref]
- Moser M, Hebert PR (1996) Prevention of disease progression, left ventricular hypertrophy and congestive heart failure in hypertension treatment trials. J Am Coll Cardiol 27: 1214-1218. [Crossref]
- Estacio RO, Jeffers BW, Hiatt WR, Biggerstaff SL, Gifford N, et al. (1998) The effect of nisoldipine as compared with enalapril on cardiovascular outcomes in patients with non-insulin-dependent diabetes and hypertension. N Engl J Med 338: 645-652. [Crossref]
- UK Prospective Diabetes Study Group (1998) Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 317: 703- [Crossref]
- Hansson L, Lindholm LH, Niskanen L, Lanke J, Hedner T, et al. (1999) Effect of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPPP) randomised trial. The Lancet 353: 611-616. [Crossref]
- Hansson L, Lindholm LH, Ekbom T, Dahlöf B, Lanke J, et al. (1999) Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity the Swedish Trial in Old Patients with Hypertension-2 study. The Lancet 354: 1751-1756. [Crossref]
- National Intervention Cooperative Study in Elderly Hypertensives (NICS-EH) Study Group (1999) Randomized double-blind comparison of a calcium antagonist and a diuretic in elderly hypertensives. Hypertens 34: 1129-1133. [Crossref]
- ALLHAT Collaborative Research Group (2000) Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). Jama 283: 1967-1975. [Crossref]
- Brown MJ, Palmer CR, Castaigne A, De Leeuw PW, Mancia G, et al.. (2000) Morbidity and mortality in patients randomised to double-blind treatment with a long-acting calcium-channel blocker or diuretic in the International Nifedipine GITS study: Intervention as a Goal in Hypertension Treatment (INSIGHT). The Lancet 356: 366-372. [Crossref]
- Hansson L, Hedner T, Lund-Johansen P, Kjeldsen SE, Lindholm LH, (2000) Randomised trial of effects of calcium antagonists compared with diuretics and β-blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) study. The Lancet 356: 359-365. [Crossref]
- Yusuf S, Sleight P, Pogue JF, Bosch J, Davies R, et al. (2000) Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 342: 145-153. [Crossref]
- Brenner BM, Cooper ME, De Zeeuw D, Keane WF, Mitch WE, et al. (2001) Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345: 861-869. [Crossref]
- Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, et al. (2002) Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. The Lancet 359: 995-1003. [Crossref]
- Black HR, Elliott WJ, Grandits G, Grambsch P, Lucente T, et al. (2003) Principal results of the controlled onset verapamil investigation of cardiovascular end points (CONVINCE) trial. Jama 289: 2073-2082. [Crossref]
- Julius S, Kjeldsen SE, Weber M, Brunner HR, Ekman S, et al. (2004) Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. The Lancet 363: 2022-2031. [Crossref]
- Rahman M, Pressel S, Davis BR, Nwachuku C, Wright JT, et al. (2004) outcomes in high-risk hypertensive patients treated with an angiotensin-converting enzyme inhibitor or a calcium channel blocker vs a diuretic: a report from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Archives of internal medicine 165: 936-946. [Crossref]
- Dahlof B, Sever PS, Poulter NR, Wedel H, Beevers DG, et al. (2005) Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. The Lancet 366: 895-906. [Crossref]
- Suzuki H, Kanno Y (2005) Effects of candesartan on cardiovascular outcomes in Japanese hypertensive patients. Hypertens Res 28: 307-314. [Crossref]
- Mochizuki S, Dahlöf B, Shimizu M, Ikewaki K, Yoshikawa M, et al. (2007) Valsartan in a Japanese population with hypertension and other cardiovascular disease (Jikei Heart Study): a randomised, open-label, blinded endpoint morbidity-mortality study. The Lancet 369: 1431-1439. [Crossref]
- Beckett NS, Peters R, Fletcher AE, Staessen JA, Liu L, et al. (2008) Treatment of hypertension in patients 80 years of age or older. N Engl J Med 358: 1887-1898. [Crossref]
- Ontarget Investigators (2008) Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 358: 1547-1559. [Crossref]
- Yusuf S, Teo K, Anderson C, Pogue J, Dyal L, Copland I, et al. (2008) Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomized controlled trial. Lancet 372: 1174-1183. [Crossref]
- Schrader J, Luders S, Kulschewski A, Hammersen F, Plate K, et al. (2005) Morbidity and mortality after stroke, eprosartan compared with nitrendipine for secondary prevention: principal results of a prospective randomized controlled study (MOSES). Stroke 36: 1218–1226. [Crossref]
- Chen GJ, Yang MS (2013) The effects of calcium channel blockers in the prevention of stroke in adults with hypertension: a meta-analysis of data from 273,543 participants in 31 randomized controlled trials. PloS one 8: e57854. [Crossref]