Loss of islet beta cell function related to inflammation is a feature of diabetes. Exposure of beta cells and islets to inflammatory mediators elevate reactive oxygen species, promote release of local factors and activate signaling pathways that lead to beta cell dysfunction. New strategies to preserve beta cell function in an inflammatory environment would be pivotal for diabetes therapy. A small compound library based on the anti-inflammatory compound Lisofylline (LSF) has been investigated to identify new molecules that confer protection to beta cells exposed to inflammatory cytokines. Efficacy was identified in compound c42. Loss of glucose-stimulated-insulin-secretion and increased beta cell apoptosis are markers of beta cell dysfunction, these were preserved with the addition of c42 to beta cell lines and primary islets. In the non-obese diabetic (NOD) mouse model of type 1 diabetes, c42 delayed the onset of hyperglycemia and reduced the frequency of diabetes by 50%. Collectively, these data support the therapeutic potential for c42 / c42-like compounds in diabetes.
Beta cell function, diabetes, inflammation, cytokines, small molecule inhibition
Loss of beta cell function is central to the etiology of diabetes. Inflammation is important in the pathophysiology of both type 1 diabetes and type 2 diabetes [1,2]. Beta cell dysfunction, beta cell apoptosis and beta cell de-differentiation are recognized pathological outcomes that result from inflammation and exposure to pro-inflammatory cytokine mediators [3-6]. Inflammatory cytokine mediators induce oxidative stress and the generation of additional local mediators that contribute to loss of functional beta cells [7-9]. Preserving beta cell function in the presence of an inflammatory environment would be a pivotal achievement in terms of diabetic therapeutics.
New biological agents to disrupt inflammatory cascades are being evaluated for diabetes patient care, and an anti-inflammatory action may be a component of existing drugs for diabetes, reviewed [10]. Transient reduction of HbA1c (a marker of sustained high blood glucose) has been reported in human type 2 diabetes following clinical trials with biological agents to disrupt interleukin-1 beta and supports the potential for inflammation disruption as a strategy to preserve beta cell function [11-13]. Lisofylline (LSF), a small molecule that is reported to disrupt inflammation by inhibition of STAT4 and /or interleukin-12 (IL-12) activation, prevents the advance of spontaneous diabetes in the type 1 diabetic NOD mouse model [14]. Th1-mediated disease (e.g. Allergic Experimental Encephalomyelitis) is suppressed by LSF through a mechanism that involves IL-12 receptor signaling blockade and inhibition of IL-12-driven Th1 differentiation and T cell proliferation [15]. Data suggest that LSF disrupts IL-12 signaling since LSF inhibits IL-12-induced STAT4 phosphorylation and prevents cytokine-mediated production of IFN-γ [16,17]. The importance of STAT4 to the development of type 1 diabetes has been shown by conferred protection to type 1 diabetes in NOD mice that are STAT4 deficient [18]. Further, polymorphism at the STAT4 locus associates with early onset type 1 diabetes in humans [19]. The potential of LSF as a therapeutic in humans is highly restricted by its negligible to low half-life. Analogs of the LSF molecule were synthesized by Cui et al creating a limited compound library [20]. Investigation of this small, but focused, LSF-like compound library for novel molecules to preserve beta cell function in an inflammatory environment has identified compound 42 (c42); an efficacious small molecule able to protect beta cells and islets from the damaging effects of inflammatory cytokines. Proof-of-concept study in the non-obese diabetic (NOD) mouse model identified compound c42 as a candidate new compound lead for the development of strategies based on small molecules that confer protection to beta cells in diabetes.
Ethics
Protocols and procedures were reviewed and approved by relevant institutional regulatory committees and comply to ARRIVE guidelines.
Cell Lines and Primary Islets
Cell lines INS-1 (rat beta cell line) and TC3 (mouse beta cell line) were cultured as previously described [21]. Human donor islets were obtained from the Integrated Islet Distribution Program (https://iidp.coh.org). At least three separate donors were used for each experimental assay. Donors were not identified as diabetic and were selected on availability only; not being matched for age, sex or BMI. Mouse islets were isolated by common bile duct cannulation and collagenase digestion [14]. Primary islets were hand picked prior to use.
Molecule Selection
Molecules from the Lisofylline-like library were resynthesized (Medchem Source, Federal Way, WA). Molecules were solubilized at 10mM in DMSO. Molecules were iteratively assessed relative to LSF. Initial screening of molecules and selection of which compounds to progress was based on rank order ability to inhibit induction of gene expression for INF γ, IL-12p35, IL-12p40, STAT4 in INS-1 cells following 4 and 24 hour exposure to pro-inflammatory cytokines (PIC: 5 ng/ml IL-1β, 10 ng/ml TNF-α, 100 ng/ml IFN-γ; R&D Systems, Minneapolis, MN). Candidate compounds were assessed at one third log concentration range that spanned 1nM to 30µM. Compound c42 was identified as a lead candidate molecule that had efficacy greater or equivalent to LSF and was subsequently evaluated in beta cell lines and primary mouse islets in biological readouts of pro-inflammatory cytokine-induced beta cell dysfunction. Compound pharmacokinetic half-life was determined in male Sprague Dawley rats following bolus intravenous injection. The analysis was conducted under contract with Frontage Laboratories, Inc. Exton, PA.
Cytokine treatment and RT PCR
Islets or cell lines were treated with species-specific pro-inflammatory cytokines (PIC: 5 ng/ml IL-1β, 10 ng/ml TNF-α, 100 ng/ml IFN-γ; R&D Systems) for 4 and 24 hours. Compound c42 was added at the stated concentration 20 min before cytokines. Isolation of RNA and quantitative RT-PCR with Taqman primers for IL-12p40, IL-12p35, IFN γ, STAT4 was performed as previously described [7,8].
STAT4 Phosphorylation
NK 92 cell nuclear extracts were prepared according to the protocol supplied with the CelLytic™ NuCLEAR™ Extraction kit (NXTRACT, Sigma-Aldrich, St. Louis, MO). Protein extracts were separated on Mini-PROTEAN® TGX™ Precast Gels (Bio-Rad, Hercules, CA) and transferred to Immobilon-FL PVDF Transfer Membranes (EMD Millipore, Billerica, MA). The blots were probed with primary antibodies and then with IRDye® 800CW and IRDye® 680RD secondary antibodies (LI-COR, Lincoln, NE) to detect protein bands with the Li-Cor Odyssey Imaging System (LI-COR). The mouse monoclonal anti-phospho-Stat4 (pY693) antibody (612739; BD Transduction Laboratories, CA) was used at 1:500. The rabbit polyclonal anti-Actin (I-19) antibody (sc-1616-R; Santa Cruz Biotechnology, Santa Cruz, CA) was used at 1:3000.
Glucose-stimulated insulin secretion (GSIS)
Cytokine treated cells or primary mouse islets were monitored for GSIS as previously described [22]. Briefly, treated cultured cells or islets were washed and stabilized in Krebs Ringer Buffer (KRB) containing 3mM glucose for one hour prior to retention in 3mM glucose or stimulation with 18 mM glucose. Ten islets were run in each condition in triplicate for each experiment. Insulin released in the culture media after 60 mins was measured by ELISA (Mercodia, Winston Salem, NC) following the manufacturer's directions.
Apoptosis detection
For caspase-3 cleavage: Apoptosis was detected using a kit to measure pro-caspase-3 cleavage (556485, BD Pharmigen, Franklin Lakes, NJ) according to the manufacturer's instructions. Conditions per experiment were performed in triplicate. Fluorescence was measured using an excitation wavelength 380 nm and emission wavelength 440 nm (SpectraMax Molecular Devices, Sunnyvale, CA).
Fluorescence microscopy was performed as reported previously [22]. All incubations were performed at least in triplicate. Briefly, following stimulation, cytokine treated cells/islets were washed with cold PBS and incubated at 4ºC for 30 min in 0.1 mM YO-PRO-1 (Y3603, Life Technologies). Five random visual fields were analyzed per well. Images were captured in 3 channels at the magnification of 100X. The fluorescence value (RFU) was normalized to background and expressed proportional to cell-occupied area (phase contrast). For quantitative analysis, area occupied by cells (obtained from the phase-contrast image channel) was determined in addition to the total densitometric value (intensity of the fluorescent signal) for the green channel (YO-PRO) within this area. For each image, the obtained densitometric value was then divided by the value obtained for the cell area and this numerical value served as a measure of apoptosis. For each experiment, the measure of apoptosis calculated for the cytokine-only treated cells was used as reference value. An apoptotic index was determined (sample normalized RFU/ cytokine treated normalized RFU x 1). Relative fluorescence expression of background normalized signal attained with cytokine-stimulation was defined as unity (maximum apoptosis). Values from other treatment conditions are expressed relative to cytokine. An Axiophot (Zeiss, Jena, Germany) was used to capture images. Axiovision (Zeiss) was used for image analysis.
NOD Mouse Model
Compound tolerability and excipient selection was first evaluated in C57BL6/J mice. NOD/ShiLtJ (NOD) female mice were purchased from The Jackson Laboratory (Bar Harbor, ME) at age 4 weeks. Mice were randomly assigned to treatment groups and dosed from age 6 weeks to age 12 weeks. Each cohort contained twenty mice that were injected intraperitoneally each weekday between 7am and 11am in alternating lower abdominal quadrants. Vehicle was 10:40:50 cremophor:PEG200:saline. Twice weekly glucose monitoring using a pre-calibrated glucometer was started at age 10 weeks. Diabetes was defined as two consecutive daily blood glucose readings ≥ 250mg/dl. Tissues including pancreas were collected fixed and paraffin embedded at study endpoints. Study was terminated at week 23.
Pancreas Histology
Sections of 7µm thickness were baked (50ºC overnight), de-paraffinized and re-hydrated in descending ethanol concentrations. Sections were subject to heat-based antigen retrieval in citrate buffer (Vector Laboratories, Burlingame, CA). Sections were incubated with 1:1000 guinea pig anti-insulin polyclonal (ab7842, Abcam, Cambridge, UK) overnight at 4ºC followed by 1:200 Cy5 conjugated secondary raised in goat (ab102372, Abcam) for 90 minutes at room temperature. Nuclei were counter stained with 0.1 µg/ml DAPI (D9542, 4',6-diamidino-2-phenylindole, Sigma, St Louis, MO) for 10 minutes. Sections were mounted with Vectashield (Vector Laboratories). All incubations were in PBS containing DAKO blocking solution, all washes between incubations were in PBS.
For each pancreas, three sections per mouse were analyzed. Whole pancreas sections were viewed, being captured in composite images at 10x objective (Axiophot, Zeiss). Images were analyzed using Axiovision software (Zeiss). Insulitis area was defined as a DAPI-visualized single structure larger in size than ten nuclei. Insulitis was evaluated by morphometry (percent of pancreatic area; sum of area of insulitis in pancreas section / total pancreas section area x 100).
Statistical Analysis
In vitro and ex vivo experiments were performed in triplicate, at minimum. Student's t-test or one-way ANOVA (Prism 5.0; Graph-Pad Software, La Jolla, CA) were used to determine statistical significance (95% CI and p < 0.05).
Study of beta cell lines and ex vivo analysis of primary islets has demonstrated the damaging effects of pro-inflammatory cytokines on beta cell function and islet survival [23,24]. Acute exposure to a highly studied triple cytokine cocktail of TNFα, IL-1β, IFNγ results in a loss of glucose-sensitive-insulin-secretion (a marker of beta cell function), enhanced oxidative stress, gene induction and elevated apoptosis [25-50]. These observations underpin the important contribution of inflammation and inflammatory mediators to diabetes etiology and identify the need for new strategies to preserve beta cell function in islets exposed to an inflammatory environment.
In an effort to identify new efficacious small molecules that uncouple the damaging effects of pro-inflammatory cytokines on beta cells, compounds from a previously developed focused molecule library of LSF-analogs were studied. This project has identified from this focused library compound c42 as a bioactive molecule that confers protection to beta cells preserving function and survival.
Selection and identification of compound c42
The small focused library of 1-(5R)-hydroxyhexyl aryl analogs were generated previously, being structural analogs of lisofylline (LSF; 1-(5R-hydroxyhexyl) 3,7-dimethylxanthine) [20]. LSF is an inhibitor of STAT4 phosphorylation that has shown beneficial effects in several inflammatory disorders [51-53]. Lisofylline prevents the advance of spontaneous diabetes in NOD mice [14]. In human islets studied ex vivo, disruption of metabolic activity and insulin secretion was reversed by LSF [54]. The clinical potential of LSF has been limited by its extremely short half-life of less than one minute in humans [20]. The rationale of the focused library of LSF-analogs was to explore chemical structure-activity-relationships beyond the xanthine framework previously investigated to determine if new efficacious molecules with different structural scaffolds could be identified [20]. Compounds were initially assessed in INS-1 rat beta cell line for ability to inhibit gene induction resulting from stimulation with pro-inflammatory cytokines (PIC). Subsequently, efficacious compounds were assessed for ability to preserve beta cell survival and function in beta cells and islets exposed to pro-inflammatory cytokines. This report describes compound 42 (c42), a molecule that shows efficacy to confer protection to beta cells exposed to pro-inflammatory cytokines. Importantly, c42 (Figure 1) does not have a xanthine-based nucleus providing new possibilities for chemical development to potentially overcome the physiochemical limitations associated with xanthine-based LSF.
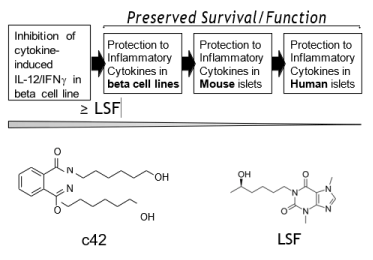
Figure 1. Compound selection. Molecules from a Lisofylline-like focused compound library were assessed for ability to block induction of IL-12 (p40, p35), INFγ and STAT4 gene expression by pro-inflammatory cytokines with molecules of interest being progressed in functional assays (flow diagram). Initial compound selection was based on greater or equi-potency to LSF in disruption of IL-12 / IFNγ gene activation by pro-inflammatory cytokines. Efficacy was subsequently assessed for candidate compounds in protection of beta cell lines, mouse islets and human islets from apoptosis induced by pro-inflammatory cytokines. Compound 42 (c42) was identified as an efficacious compound. Molecular structure of c42 and LSF are shown.
Preservation of beta cell function in beta cell lines exposed to PICs
Pro-inflammatory cytokine (PIC) exposure of rat beta cell line, INS-1, results in loss of glucose-stimulated-insulin-secretion, elevated expression of IL-12 genes (p40 and p35) and induction of apoptosis. The IL-12/IFNγ stimulatory axis is induced in islets as a response to pro-inflammatory cytokine stimulation and is linked to beta cell dysfunction [7, 16]. Following stimulation of INS-1 cells with PIC, gene expression for IL-12p40, IL-12p35, IFNγ and STAT4 elevated significantly above control, vehicle treated cells (3.75±0.9, 2.53±0.8, 7.67±2.4 and 7.32±2.0 fold respectively). In the presence of c42 the PIC-induced gene expression was inhibited by 66%±8, 57%±10, 52%±15 and 43%±4 for IL-12p40 (p<0.01), IL-12p35 (p<0.01), IFN (p<0.05) and STAT4 (p<0.05), respectively (Figure 2A). Beta cells exposed to an elevation of glucose concentration release insulin in culture. Glucose-stimulated-insulin-secretion (GSIS) is uncoupled with exposure to PICs providing a sensitive marker of beta cell dysfunction. The addition of c42 along with PICs protected the GSIS response, demonstrating preserved beta cell function (Figure 2B, p<0.05). Exposure of beta cells and islets to PICs also elevates cell death. Caspase 3 activation or incorporation of the fluorescent dye YO-PRO-1 are markers of cell apoptosis and are elevated in beta cells following treatment with pro-inflammatory cytokines. The addition of 20µM c42 inhibited apoptosis induced by PICs (Figure 2C-D, p<0.01 and p<0.0001 respectively). The protective effect of c42 to beta cells is shown relative to 20µM LSF (Figure 2B-D).
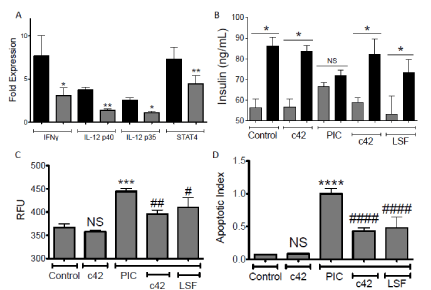
Figure 2. Induced beta-cell dysfunction in rat pancreatic INS-1 cells is inhibited by compound c42. Inhibition of PIC-induced gene induction (A), protection to PIC-induced uncoupling of GSIS (B), or blockade of PIC induced apoptosis (C, caspase3) and (D, YO-PRO-1) by 20µM c42 or LSF (B-D) or c42 (A) is shown in INS-1 cells. Fold expression by PICs is shown in the absence (black bar) or presence (grey bar) of 20µM c42 (A). For GSIS (B), induced insulin secretion is compared between 3mM glucose (grey bars) and 18mM glucose (black bars). Graphs (C,D) display caspase activation RFU (relative fluorescent units) and fluorescent apoptotic index (YO-PRO-1) respectively. Compound c42 on its own had no significant (NS) effect (B,C,D). ****, ***, **, *; p<0.0001, <0.001, <0.01, <0.05, to Control respectively; ####, ##, #; p<0.0001, <0.01, <0.05, to PIC treatment respectively. Data within each experiment is from triplicate replicates and results shown are combined data from three or greater separate experiments.
Compound c42 demonstrated efficacy analogous to LSF in its ability to preserve beta cell function and survival. Compound c42 is equipotent to LSF. The extremely short pharmacokinetic half-life of LSF is a major limitation to its clinical utilization. Preliminary pharmacokinetic analysis of c42 in Sprague-Dawley rats following intravenous administration determined a calculated half-life of forty-five minutes. In contrast, reported half-life for LSF is seven minutes [55]. Further medicinal chemical refinement of c42 may enhance the half-life of c42. However, in vivo efficacy is demonstrated with systemic administration of c42.
Compound-driven protection in mouse beta cells and primary islets from PIC-induced apoptosis
The ability of c42 to protect a mouse beta cell line and primary isolated islets from apoptosis that is induced by exposure to pro inflammatory cytokines (TNFα, IL-1β, IFNγ) was assessed. The mouse beta cell line βTC3, primary isolated mouse islets and primary human donor islets were studied (Figure 3). Apoptosis was determined through two quantitative methodologies, incorporation of the cellular fluorescence dye YO-PRO-1 and conversion of a Caspase-3 substrate, as described [22]. Exposure of beta cells and islets to PICs results in a significant increase in apoptosis relative to vehicle control (p<0.01). In the presence of c42 a significant inhibition of PIC-induced apoptosis is seen in mouse beta cells, mouse islets and human donor islets (Figures 3A and 3C, p<0.001). To provide further evidence that c42 confers protection to beta cells and islets from PIC-induced apoptosis, induction of caspase-3 was assessed in mouse cells. Both mouse beta cell lines and primary isolated mouse islets show a significant increase in caspase activity following exposure to PICs (Figures 3D and 3E, p<0.05). The induction in caspase activity was inhibited with the inclusion of c42 (p<0.01). These data show that c42 confers a survival advantage to beta cells and islets exposed to pro inflammatory cytokines and raise the possibility of efficacy to protect beta cells against inflammation and inflammatory mediators that are present in the development of diabetes.
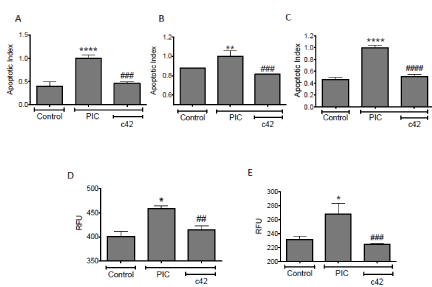
Figure 3. Induced beta-cell apoptosis in pancreatic islets and beta cells is inhibited by compound c42. Apoptosis, measured by YO-PRO-1 (A-C) and caspase-3 (D, E), in TC-3 beta cells (A, D), primary mouse islets (B, E), and primary human donor islets (C) treated with PICs in the absence or presence of 50µM c42. Graphs display fluorescent apoptotic index (YO-PRO-1) (A –C) and caspase activation RFU (relative fluorescent units, D, E). ****, **, *; p<0.0001, <0.01, <0.05, to Control respectively; ####, ###, ## p<0.0001, <0.001, <0.01, to PIC treatment respectively. Data within each experiment is from triplicate replicates and results shown are combined data from three or more separate experiments.
Disruption of type 1 diabetes development in NOD mice by c42
To provide proof-of-concept efficacy in a diabetes model, assessment was made using the non-obese diabetic (NOD) mouse model. The female NOD mouse is an inbred strain that spontaneously develops islet inflammation (insulitis), beta cell destruction and hyperglycemia [56]. Dosing NOD mice with c42 prior to the development of insulitis provided a proof-of-concept model to evaluate the efficacy of c42 in delaying the onset or reducing the incidence of type 1 diabetes in this mouse model.
Evaluation of a range of excipients determined that the vehicle Cremophor EL:PEG 200:Saline (10:40:50) effectively solubilized c42 and this vehicle was tolerated by mice in repeat daily intraperitoneal injections with no adverse effects. To evaluate c42 in an initial study of in vivo efficacy, NOD mice were injected by intraperitoneal route from age 6 weeks for six consecutive weeks. The two injected groups were vehicle alone and vehicle containing c42 at 28mg/Kg. Each cohort had twenty mice. Blood glucose in the mice was monitored twice weekly starting at week ten. Hyperglycemia (conversion to diabetes) was defined as two consecutive daily blood glucose readings at or above 250 mg/dl. These data were compared to our institutional diabetic conversion rate for non-treated NOD mice. The rate of diabetes conversion for NOD mice, even when sourced from the same vendor’s colony, varies between institutions being dictated by environment. It is important to make comparison to the established institutional conversion rate/ time frame. Mice treated with c42 had a delayed initiation of diabetes development (15 weeks c42 dosed vs 10/12 weeks vehicle control) and a lower conversion to diabetes at week 23 occurred in c42 dosed mice when compared to vehicle control or the non-treated group (Figure 4A, 31% vs 54 or 52% resp.). The slopes for diabetic conversion was significantly different (p<0.001) in the c42 group (Figure 4B), slopes were 3.33±0.40, 4.86±0.33 and 4.62± 0.28 for c42 treated, vehicle control and non-treated respectively. Representative mice from each group were assessed at age 12 weeks, the remaining mice were progressed to study endpoint. Pancreata were recovered and analyzed for insulitis (presence of inflammatory cells around islets) by histology (Figure 5). A significant inhibition in insulitis was observed at age 12 weeks in mice treated with c42 when compared to vehicle control (p<0.0001 age 12 weeks). Representative images of islets are shown (Figure 5B). At age 12 weeks mice had been dosed for 6 weeks prior with compound or vehicle. Pancreas sections were co-stained for insulin to identify beta cells. Predominantly, islets in the vehicle control group that were surrounded by inflammatory cells presented with reduced insulin positive cells (Figure 5B a-c). In contrast, islets from c42 treated mice had preserved insulin positive cells. (Figure 5B d-f). These observations of insulitis and insulin at week 12, while not on their own conclusive, corroborate the suppressed development of hyperglycemia in the c42 treated mice group. Analysis of NOD mice pancreata at study endpoint showed a significantly higher area of insulin positive cells in the c42 treated group relative to vehicle (p<0.05, Figure 5C).
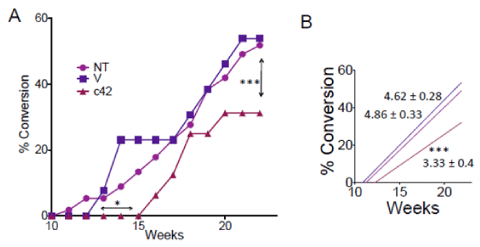
Figure 4. Inhibition of conversion to diabetes by compound c42 in NOD mice. Development of diabetes (blood glucose ≥ 250mg/dl) in NOD mice is shown in A for groups; 28mg/Kg compound c42 (c42, triangle), untreated control (NT, circle) and vehicle alone (V, square). Arrows (double headed) mark delay in diabetes onset by c42 (p<0.01) and decrease conversion to diabetes by c42 at age 23 weeks (p<0.001). Linear regression line for each group (B) shows significant difference in slope for c42 treated group relative to controls (V, NT, color coded) (p<0.01).
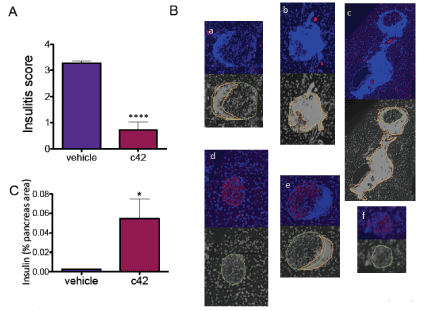
Figure 5. Reduced islet inflammation following treatment with compound c42 in NOD mice. Insulitis score in pancreas from vehicle and c42 treated mice at age 12 weeks (A, p<0.0001). Representative islet images are shown (B) for and vehicle treated (a-c) and compound c42 treated (d-f) NOD mice at age 12 weeks. Pancreatic sections were stained for DAPI (blue) to mark cell nuclei and insulin (red) to mark beta cells. Insulitis shows as dense infiltrate of DAPI positive cells in the peri-islet region. Monochrome images outline islet (yellow dashed line) and insulitis (orange line). Islets in vehicle control mice are predominantly surrounded by inflammatory infiltrate and present with diminished or negligible insulin stain (erythrocytes that autofluoresce are seen in blood vessels as small red spots). Insulin stain is preserved in islets from c42 treated mice. Insulin positive area in the pancreata of mice at study endpoint is graphed (C) for vehicle group and C42 treated group. *, **** p<0.05, 0.0001 respectively.
Collectively, the data show protection to diabetes conversion in the NOD model by c42. A significant reduction in inflammatory cells in the pancreas and preservation of insulin expressing beta cells, further support the efficacy of c42. These proof-of-concept data evolve the in vitro/ex vivo protection that c42 treatment provides to beta cells and islets exposed to inflammatory cytokines. The islet protection demonstrated by c42 is encouraging particularly considering the study mice had limited exposure to the molecule. Six weeks of weekday single dose compound administration may not be optimum. Follow up investigations with more sustained compound exposure is justified. It is noteworthy that mice dosed with c42 for 6 weeks exhibited no adverse events.
The mechanism of action for c42 is not fully characterized requiring further investigation. Based on an analogous inhibition of IFNγ and IL-12 gene expression, c42 is predicted to have a similar mechanism of action to the parent compound LSF. However, while c42 clearly disrupts IL-12 activation, the possibility remains that the target for inhibition by c42 may be distinct to LSF. The presence of phosphorylated STAT4 in the nucleus of human Natural Killer cell line (NK92) following stimulation with IL-12 while being significantly inhibited by LSF was not modified by c42 (Supplemental Figure). Additional study on the molecular inhibition profile and mechanism of inhibition of c42 to accompany its clear biologic efficacy in protection / preservation of beta cells is warranted.
In conclusion, this study identifies a promising new molecule to aid the development of new therapeutic approaches for diabetes and enhance the discovery of additional efficacious compounds to reduce the negative effects of pro inflammatory cytokines on beta cell function/survival. The potential of c42, or future c42-like compounds, is highlighted by proof-of-principle efficacy of c42 in the NOD mouse model.
The research was supported by grants to DATF from Department of Defense (PR093521) and Commonwealth Research Commercialization Fund (MF13-029-LS). The valued contributions to the mice study by Rebekah Raab and Kendall Leone are recognized. Andrew Pearson helped in figure preparation.
- Donath MY (2014) Targeting inflammation in the treatment of type 2 diabetes: time to start, Nature reviews. Drug discovery 13: 465-476.
- Soleimanpour SA, Stoffers DA (2013) The pancreatic β cell and type 1 diabetes: innocent bystander or active participant? Trends Endocrinol Metab 24: 324-331. [Crossref]
- Nordmann TM, Dror E, Schulze F, Traub S, Berishvili E, et al. (2017) The Role of Inflammation in β-cell Dedifferentiation. Sci Rep 7: 6285. [Crossref]
- Negi S, Jetha A, Aikin R, Hasilo C, Sladek R, Paraskevas S (2012) Analysis of beta-cell gene expression reveals inflammatory signaling and evidence of dedifferentiation following human islet isolation and culture PloS one 7: e30415.
- Hasnain SZ, Prins JB, McGuckin MA (2016) Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes. J Mol Endocrinol 56: R33-54. [Crossref]
- Berchtold LA, Prause M, Størling J, Mandrup-Poulsen T (2016) Cytokines and Pancreatic β-Cell Apoptosis. Adv Clin Chem 75: 99-158. [Crossref]
- Taylor-Fishwick DA, Weaver JR, Grzesik W, Chakrabarti S, Green-Mitchell S, et al. (2013) Production and function of IL-12 in islets and beta cells. Diabetologia 56: 126-135. [Crossref]
- Weaver JR, Holman TR, Imai Y, Jadhav A, Kenyon V, et al. (2012) Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction Mol Cell Endocrinol 358: 88-95.
- Clark AL, Kanekura K, Lavagnino Z, Spears LD, Abreu D, et al. (2017) Targeting Cellular Calcium Homeostasis to Prevent Cytokine-Mediated Beta Cell Death, Sci Rep 7 5611.
- Pollack RM, Donath MY, LeRoith D, Leibowitz G (2016) Anti-inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications Diabetes Care 39: S244-S252.
- Peiro C, Lorenzo O, Carraro R, Sanchez-Ferrer CF (2017) IL-1beta Inhibition in Cardiovascular Complications Associated to Diabetes Mellitus Front Pharmacol 8: 363.
- Sloan-Lancaster J, Abu-Raddad E, Polzer J, Miller JW, Scherer JC, et al. (2013) Double-blind, randomized study evaluating the glycemic and anti-inflammatory effects of subcutaneous LY2189102, a neutralizing IL-1beta antibody, in patients with type 2 diabetes Diabetes care 36: 2239-2246.
- Hensen J, Howard CP, Walter V, Thuren T (2013) Impact of interleukin-1beta antibody (canakinumab) on glycaemic indicators in patients with type 2 diabetes mellitus: results of secondary endpoints from a randomized, placebo-controlled trial Diabetes Metab 39: 524-531.
- Yang ZD, Chen M, Wu R, McDuffie M, Nadler JL (2002) The anti-inflammatory compound lisofylline prevents Type I diabetes in non-obese diabetic mice. Diabetologia 45: 1307-1314. [Crossref]
- Bright JJ, Du C, Coon M, Sriram S, Klaus SJ (1998) Prevention of experimental allergic encephalomyelitis via inhibition of IL-12 signaling and IL-12 mediated Th1 differentiation: an effect of the novel anti-inflammatory drug lisofylline. J Immunol 161: 7015-7022.
- Weaver JR, Nadler JL, Taylor-Fishwick DA (2015) Interleukin-12 (IL-12)/STAT4 Axis Is an Important Element for beta-Cell Dysfunction Induced by Inflammatory Cytokines PloS one 10: e0142735.
- Coon ME, Diegel M, Leshinsky N, Klaus SJ (1999) Selective pharmacologic inhibition of murine and human IL-12-dependent Th1 differentiation and IL-12 signaling. J Immunol 163: 6567-6574.
- Yang Z, Chen M, Ellett JD, Fialkow LB, Carter JD, et al. (2004) Autoimmune diabetes is blocked in Stat4-deficient mice. J Autoimmun 22: 191-200. [Crossref]
- Lee HS, Park H, Yang S, Kim D, Park Y (2008) STAT4 polymorphism is associated with early-onset type 1 diabetes, but not with late-onset type 1 diabetes. Ann N Y Acad Sci 1150: 93-98. [Crossref]
- Cui P, Macdonald TL, Chen M, Nadler JL (2006) Synthesis and biological evaluation of lisofylline (LSF) analogs as a potential treatment for Type 1 diabetes, Bioorg Med Chem Lett 16: 3401-3405.
- Scharfmann R, Tazi A, Polak M, Kanaka C, Czernichow P (1993) Expression of functional nerve growth factor receptors in pancreatic beta-cell lines and fetal rat islets in primary culture Diabetes 42: 1829-1836.
- Weaver JR, Grzesik W, Taylor-Fishwick DA (2015) Inhibition of NADPH oxidase-1 preserves beta cell function. Diabetologia 58: 113-121. [Crossref]
- Cieślak M, Wojtczak A, Cieślak M (2015) Role of pro-inflammatory cytokines of pancreatic islets and prospects of elaboration of new methods for the diabetes treatment. Acta Biochim Pol 62: 15-21. [Crossref]
- Lambelet M, Terra LF, M Fukaya K, Meyerovich L, Labriola AK, et al. (2018) Dysfunctional autophagy following exposure to pro-inflammatory cytokines contributes to pancreatic beta-cell apoptosis Cell Death Dis 9: 10-96.
- Rabinovitch (1998) An update on cytokines in the pathogenesis of insulin-dependent diabetes mellitus, Diabetes Metab Rev, 14:129-151.
- Imai Y, Dobrian AD, Morris MA, Taylor-Fishwick DA, Nadler JL (2016) Lipids and immunoinflammatory pathways of beta cell destruction Diabetologia 59: 673-678.
- Mandrup Poulsen T, Bendtzen K, Nielsen JH, Bendixen G, Nerup J (1958) Cytokines cause functional and structural damage to isolated islets of Langerhans Allergy 40: 424-429.
- Rabinovitch A, Sumoski W, Rajotte RV, Warnock GL (1990) Cytotoxic effects of cytokines on human pancreatic islet cells in monolayer culture J Clin Endocrinol Metab 71: 152-156.
- Grunnet LG, Aikin R, Tonnesen TF, Paraskevas S, Blaabjerg L, et al. (2009) Mandrup-Poulsen, Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells, Diabetes 58: 1807-1815.
- Allagnat F, Fukaya M, Nogueira TC, Delaroche D, Welsh N, et al. (2012) C/EBP homologous protein contributes to cytokine-induced pro-inflammatory responses and apoptosis in beta-cells Cell Death Differ 19: 1836-1846.
- Burke SJ, Goff MR, Updegraff BL, Lu D, Brown PL, et al. (2012) Regulation of the CCL2 gene in pancreatic β-cells by IL-1β and glucocorticoids: role of MKP-1. PLoS One 7: e46986. [Crossref]
- Dahllof MS, Christensen DP, Lundh M, Dinarello CA, Mascagni P, et al. (2012) The lysine deacetylase inhibitor Givinostat inhibits beta-cell IL-1beta induced IL-1beta transcription and processing, Islets 4: 417-422.
- Hansen JB, Tonnesen MF, Madsen AN, Hagedorn PH, Friberg J, et al. (2012) Mandrup-Poulsen, Divalent metal transporter 1 regulates iron-mediated ROS and pancreatic beta cell fate in response to cytokines Cell Metab, 16: 449-461.
- Stojanovic I, Saksida T, Nikolic I, Nicoletti F, Stosic Grujicic I (2012) Macrophage migration inhibitory factor deficiency protects pancreatic islets from cytokine-induced apoptosis in vitro Clin Exp Immunol 169: 156-163.
- Ngamjariyawat A, Turpaev K, Vasylovska S, Kozlova EN, Welsh N (2013) Co-culture of neural crest stem cells (NCSC) and insulin producing beta-TC6 cells results in cadherin junctions and protection against cytokine-induced beta-cell death, PloS one 8: e61828.
- Lortz S, Schroter S, Stuckemann V, Mehmeti I, Lenzen S (2014) Influence of cytokines on Dmt1 iron transporter and ferritin expression in insulin-secreting cells. J Mol Endocrinol 52: 301-310.
- Pepin E, Higa A, Schuster-Klein C, Bernard C, Sulpice T, et al. (2014) Deletion of apoptosis signal-regulating kinase 1 (ASK1) protects pancreatic beta-cells from stress-induced death but not from glucose homeostasis alterations under pro-inflammatory conditions, PloS one 9: e112714.
- Verma MK, Sadasivuni MK, Yateesh AN, Neelima K, Mrudula S, et al. (2014) Activation of GPR40 attenuates chronic inflammation induced impact on pancreatic beta-cells health and function, BMC Cell Biol 15: 24.
- Zhu Q, Kang J, Miao H, Feng Y, Xiao L, et al. (2014) Low-dose cytokine-induced neutral ceramidase secretion from INS-1 cells via exosomes and its anti-apoptotic effect, FEBS J 281: 2861-2870.
- Brandhorst H, Theisinger B, Guenther B, Johnson PR, Brandhorst D (2015) Pancreatic L-Glutamine Administration Protects Pig Islets from Cold Ischemic Injury and Increases Resistance toward Inflammatory Mediators Cell Transplant.
- Burke SJ, Stadler K, Lu D, Gleason EL, Han A, et al. (2015) IL-1beta reciprocally regulates chemokine and insulin secretion in pancreatic beta-cells via NF-kappaB, American journal of physiology. Endocrinology and metabolism
- Chan Y, Luzuriaga J, Maxwell EL, West PK, Bensellam M, et al. (2015) The balance between adaptive and apoptotic unfolded protein responses regulates beta-cell death under ER stress conditions through XBP1, CHOP and JNK. Mol Cell Endocrinol 413: 189-201.
- Gorasia DG, Dudek NL, Veith PD, Shankar R, Safavi-Hemami H, et al. (2015) Purcell, Pancreatic beta cells are highly susceptible to oxidative and ER stresses during the development of diabetes, J Proteome Res 14: 688-699.
- Ibarra Urizar A, Friberg J, Christensen DP, Lund Christensen G, Billestrup N (2015) Inflammatory Cytokines Stimulate Bone Morphogenetic Protein-2 Expression and Release from Pancreatic Beta Cells. J Interferon Cytokine Res.
- Lortz S, Lenzen S, Mehmeti I (2015) Impact of scavenging hydrogen peroxide in the endoplasmic reticulum for β cell function. J Mol Endocrinol 55: 21-29. [Crossref]
- Paula PM, Leite NC, Vanzela EC, Kurauti EA, Freitas-Dias R, et al. (2015) Exercise increases pancreatic beta-cell viability in a model of type 1 diabetes through IL-6 signaling, FASEB J 29: 1805-1816.
- Shahbazov R, Kanak MA, Takita M, Kunnathodi F, Khan O, et al. (2015) Essential phospholipids prevent islet damage induced by proinflammatory cytokines and hypoxic conditions. Diabetes Metab Res Rev.
- Turpaev K, Welsh N (2015) Brusatol inhibits the response of cultured beta-cells to pro-inflammatory cytokines in vitro. Biochem Biophys Res Commun 460: 868-872. [Crossref]
- Xiang RL, Mei M, Su YC, Li L, Wang JY (2015) Visfatin Protects Rat Pancreatic beta-cells against IFN-gamma-Induced Apoptosis through AMPK and ERK1/2 Signaling Pathways Biomed Environ Sci 28: 169-177.
- Zheng Y, Wang Z, Tu Y, Shen H, Dai Z, et al. (2015) miR-101a and miR-30b contribute to inflammatory cytokine-mediated β-cell dysfunction. Lab Invest 95: 1387-1397.
- Chen M, Yang Z, Wu R, Nadler JL (2002) Lisofylline, a novel antiinflammatory agent, prevents pancreatic cells from proinflammatory cytokine damage by promoting mitochondrial metabolism, Endocrinology 143: 2341-2348.
- Rice GC, Brown PA, Nelson RJ, Bianco JA, Singer JW, et al. (1994) Protection from endotoxic shock in mice by pharmacologic inhibition of phosphatidic acid. Proc Natl Acad Sci U S A 91: 3857-3861. [Crossref]
- Wattanasirichaigoon S, Menconi MJ, Fink MP (2000) Lisofylline ameliorates intestinal and hepatic injury induced by hemorrhage and resuscitation in rats. Crit Care Med 28: 1540-1549. [Crossref]
- Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick RA (2010) Nadler, 12-Lipoxygenase Products Reduce Insulin Secretion and {beta}-Cell Viability in Human Islets, J Clin Endocrinol Metab 95: 887-893.
- Weaver JR, Nadler JL, Taylor-Fishwick RA (2015) Interleukin-12 (IL-12)/STAT4 Axis Is an Important Element for beta-Cell Dysfunction Induced by Inflammatory Cytokines, PloS one 10: e0142735.
- Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, et al. (1980) Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu 29: 1-13. [Crossref]