IgE production is a unique characteristic of mammals. Traditionally, the concept of allergy implied an abnormal response to an otherwise benign agent (eg, pollen or food), with an easily identifiable relationship between exposure and disease. However, there are syndromes in which the relationship between exposure to the relevant allergen and the "allergic" disease is not clear. In these cases, the presence of specific IgE antibodies can play an important role in identifying the relevant allergen and provide a guide to therapy. IgE is the antibody isotype found at the lowest concentration in the circulation. However, IgE can undeniably play an important role in mediating allergic reactions; best exemplified by the clinical benefits of anti-IgE monoclonal antibodies therapy for some allergic diseases. IgE has also been implicated in congenital diseases due to specific molecular defects and in autoimmune diseases behaving as a self-reactive immunoglobulin. This review will describe our current understanding of the interactions between IgE and its main receptors FcεRI and CD23 (FcεRII). We will review the known and potential functions of IgE in health and disease: in particular, its detrimental roles in allergic diseases and, its protective functions in host defense against parasites and venoms. Finally, we will present an overview of the drugs that are in clinical development or have therapeutic potential for IgE-mediated allergic diseases.
allergy, IgE, autoimmunity, monoclonal antibodies, atopy
The allergic phenotype has likely saved the lives of many more mammals than have ever died from allergy, so justifying the positive role of IgE in our evolution. The resultant type I hypersensitivity affects a large section of the world's population, with those, reporting allergy steadily increasing from the 1960s and 1970s. Today, an estimated 40% of the population are sensitized (ie have specific IgE) to environmental proteins [1]. Therefore, IgE‐dependent type I hypersensitivity remains medically problematic. The allergic phenotype, despite its apparent high cost, offers evolutionary value through a range of benefits. These range from protection against parasitic infection for a large proportion of the world's population, to accelerated wound healing, protection against venoms and possibly tumours. That IgE occurred early during evolution of mammals, probably due to an IgY gene duplication event, is supported by the observation that this immunoglobulin occurs in all mammals [2]. The structure of IgE appears well conserved throughout mammals, pointing to strong evolutionary pressures to maintain its full functionality intact [3]. Phylogenetic evidence suggests that the IgE receptor evolved from Fc receptor‐like (FcRL) molecules early during mammalian evolution [4]. Mast cells and basophils, in all, likelihood precede the advent of both IgE and its high affinity receptor, as does the protease content of their granules. Apparently the first allergens originated from pollens, spores and insects that were part of the diet of ancestral mammals, 360 million years ago. The elderly allergens of protozoa and helminths also date from the same times, in such a way that all the ingredients for the IgE-mediated allergic response existed with the appearance of mammals. Namely: mast cells and basophils, allergens, Ig-E, and its receptors, and possibly Th2 cytokines [5].
Immunoglobulin E (IgE) was discovered about 54 years ago. In 1966, the Ishizakas' group in Japan described an immunoglobulin differ from the known immunoglobulin classes, that could induce allergic reactions in the skin, and which they called γE antibody [6]. The official name IgE was given in 1968. IgE is the isotype found at the lowest concentration in the circulation (50–200 ng/ml IgE in healthy individuals vs.~10 mg/ml for IgG). However, IgE levels can increase dramatically in individuals with allergic diseases [7]. This discovery has had great importance for both the diagnosis and treatment of allergic disorders: quantification of allergen specific IgE is one of the main diagnostic criteria for allergies [8]. IgE antibodies exist in two forms: a membrane-bound form (mIgE) expressed by B cells that have undergone class switching to IgE, and a secreted form produced by plasma B cells. mIgE serves as a B cell receptor involved in antigen uptake and presentation. The half-life of serum IgE is 3 days but tissue IgE is weeks and even months. IgE exerts its biological functions by binding to two main receptors: FcεRI and CD23 (FcεRII). The high affinity IgE receptor, FcꞓRI, is expressed on the surface of blood basophils and tissue resident mast cells; and on other cell types in humans, including neutrophils, eosinophils, platelets, monocytes, and dendritic cells [9]. Low affinity receptor CD23 is expressed mainly by B cells [10], but also by several other cell populations including neutrophils, eosinophils, follicular DCs and intestinal epithelial cells (IECs). CD23 on B cells serves mainly as a negative regulator of IgE synthesis.
IgE is a glycoprotein monomeric antibody. IgE antibodies are composed of two identical heavy chains (each comprising a variable VH domain and four constant Cε domains) and two identical light chains (composed of a variable VL domain and a constant CL domain) with a total molecular weight of 190 kDa (Figure 1). Similar, to other antibody classes, the Fab region of IgE is responsible for antigen recognition and binding, while the effector function of IgE is determined by the carboxy-terminal Fc portion [11]. The FcεRI binding site is located, in the Cε3 domain and in the Cε2-Cε3 linker region. The binding site to the low affinity IgE receptor CD23 is also primarily located within the Cε3 domain, with contributions from the Cε4 domain. The crystal structure of the human Cε3-Cε4 domains revealed that, by rotating relatively to Cε4, Cε3 can adopt either ‘open’ or ‘closed’ conformations. This conformational flexibility regulates the binding of IgE to both FcεRI and CD23. Several intra- and inter-domain disulphide bridges control the structure and activity of IgE. The activity of IgE is also regulated by glycosylation at various sites [9].
Figure 1. IgE structure
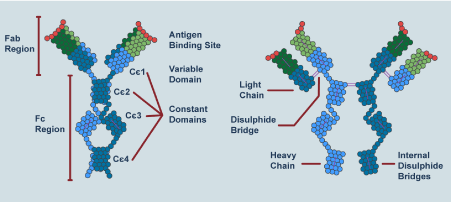
IgE antibodies consist of two identical heavy chains (composed of a variable VH domain and four constant Cε domains) and two identical light chains (composed of a variable VL domain and a constant CL domain). ‘Fab’: region responsible for antigen recognition and binding. ‘Fc’: portion responsible for IgE effector functions. The positions of interdomain disulfide bridges are indicated.
FcεRI is the high affinity receptor for IgE (Kd of ~10−9 to 10−10 M) (Kd=dissociation constant). The receptor is a tetramer that is expressed at high levels predominantly in basophils and mast cells, and has an α and a β subunit, and Ɣ dimer linked by disulfide bridges. There is a circulating form of the receptor whose physiological role is yet to be identified. The cell-bound receptor is the one that mediates biological functions. CD23, also known as FcεRII, is the low affinity receptor for IgE (Kd = 10−5 M). The receptor is a trimer and is preferentially expressed on the surface of B cells and has been involved in antigen uptake and presentation (IgE-dependent) to T cells. There is also a soluble form of this receptor [12].
The classic Gell and Coombs classification defines 4 types of immune reactions. The type I immediate hypersensitivity reaction, mediated by IgE, which is the one addressed in this monograph (eg, anaphylaxis). Type II cytotoxic hypersensitivity reaction mediated by IgG or IgM but bound by such antibodies to cell surface antigens (eg, drug-induced hemolytic anemia). The type III reaction produced by circulating immune complexes that then bind to postcapillary venules (eg, serum sickness) and the type IV reaction, which is a delayed hypersensitivity reaction, mediated not by antibodies but by cells (eg, PPD reaction in Tuberculosis). Other classifications add up to 3 more types of reactions but the Gell and Coombs classification is practical and academic. IgE is also known as "reagin" because that is how it was conceptualized when the “reaction” it produced was described. Some immediate hypersensitivity reactions may not be mediated by IgE but by IgG4 [13]. Allergy is a hypersensitivity reaction mediated by an immune mechanism that involves antibodies or cells. Atopy is a family or personal tendency to produce IgE-like antibodies in response to low doses of antigens, usually proteins. An "anaphylactoid" reaction is a reaction similar, to an anaphylactic reaction but not mediated by IgE (non-IgE–mediated reactions). The genes that encode the synthesis of IgE are located on chromosome 14 [13].
It is a type of immunity that provide protection against parasitic infections but is also active in allergic diseases. It is orchestrated by the CD4+ T helper 2 (Th2] cells and the innate lymphoid cells group 2 (ILC2s) through the secretion of IL-4, IL-5, IL-9, and IL-13, which are regulated by the transcription factor GATA3. GATA3 is a transcription factor that in humans is encoded by the GATA3 gene, GATA is one of three genes muted > 10% of breast cancer (Cancer Genome Atlas). In the type I immediate hypersensitivity reaction the cascade of events is as follows during allergen sensitization. The epithelial cells, in response to allergens, will release cytokines such alarmins (IL-25, IL-33 and TSLP [thymic stromal lymphopoietin]) which program the immature DCs (dendritic cells) of the respiratory tract to process the allergens and migrate to the local lymph nodes, present the peptides resulting from the processing to the naive or uncommitted T lymphocytes and generate Th2.
DCs are specialized cells similar macrophages, that are in the epithelium of airway and are the major line of antigen presenting cells and mature when presenting the antigens coupled to the major histocompatibility complex II (MHC II) to the T system. DCs activate and recruit Th2, through CCL17 and CCL 22 (CC-chemokines ligand 17 and 22 respectively), but these chemokines can directly activate ILC2s cells present in the mucosa of the airway. The Th2, through the activation of the transcription factor STAT 6 (signal transducer and activator of transcription) and the ILCs2, through the activation of GATA3, secrete Th2 cytokines (Figure 2) [14]. These interleukins will lead to airway infiltration with eosinophils (IL-5], B cells (IL-4 e IL-13], which become plasma cells and generate IgE in response to allergens and alternatively these interleukins activated macrophages, which will produce obstruction and symptoms, and IL-9, attracts and activates mast cell proliferation.
Figure 2. Type 2 immunity
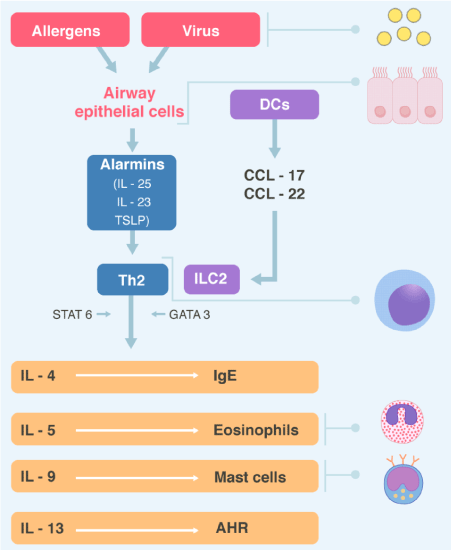
Virus and inhaled allergens activated lung dendritic cells (DCs) to release the CC chemokines ligand 17 (CCL17) and CCL22 which recruit T helper 2 (Th2) cells and group innate lymphoid cells (ILC2s). Activation of the transcription factor GATA3 and STAT6 in Th2 cells and ILC2s leads to secret of the cytokines IL-4, IL-5, IL-9, IL-13. These cytokines regulated IgE synthesis, eosinophilic proliferation, mast cells proliferation, and airway hyperreactivity (AHR) respectively. Epithelial cells also producing alarmins, including thymic stromal lymphopoietin (TSLP), Il-25 and IL-33, which are “upstream” cytokines in the eosinophilic response. STAT6= Signal transduction and activator of transcription 6.
Specifically, with respect to the control and production of IgE, two aspects deserve to be highlighted. First, THF (follicular helper T) rather than Th2 appear to be responsible for orchestrate the IgE production. TFH are located on the B cell follicles in the secondary lymphoid organs. Upregulation of TFH have been associated with allergic diseases. Second, IgE production requires 2 communication signals between T and B cells to convert Ig G to IgM, and IgM to IgE, in plasma cells. The first signal is the one discussed since it involves IL-4 and IL-13 and the second is the interface between CD40 B cells and the CD40 ligand of T cells (CD 154] [15,16]. This occurs due to an immunoglobulin isotype change that involves somatic recombination mediated by AID (activation induced by cytosine deaminase) [17]. IgE binds to its high and low affinity receptors in airways mast cells, circulating basophils and other mucosal cells and when re-exposure to the allergen occurs, they will release mediators already preformed in their granules such as histamine, proteases (hydrolases, chymases and carboxypeptidases), lipid mediators formed "de novo" (leukotrienes, prostaglandins and thromboxanes), cytokines and chemokines, which will damage the airway and amplify the inflammatory process [18].
The other cells that infiltrate the airway in the type I reaction are eosinophils. Attracted from blood and bone marrow by CCL11 (eotaxin), CCL5 (RANTES = regulated upon activation, normal T cell expressed and secreted) and IL-5 will release their four major proteins (basic major protein, the eosinophil cation protein, the eosinophil-peroxidase, and the eosinophil-derived neurotoxin), which they are capable of inducing tissue damage and dysfunction [19].
Therefore, the cellular profile that infiltrates the airway (or any other tissue that is the target of a type I reaction such as skin, nasal, or gastrointestinal mucosa) produced by the Th2 program is made up of mast cells, eosinophils, and Th2 lymphocytes.
Allergic diseases
Bronchial asthma is a chronic and common inflammatory respiratory disease that affects 10% of the adult population in different countries [20]. An estimated 315 million affected people worldwide and 346,000 deaths every year. It is more difficult to assess the prevalence in children under 5 years old for several reasons. It is difficult to make a reliable diagnosis of asthma in this age group because respiratory findings such as cough and bronchospasm are frequent in children without asthma, particularly in those 0-2 years of age. Some viral infections (respiratory syncytial and rhinovirus) are associated with recurrent wheezing throughout childhood. Therefore, not all wheezing in this age means asthma. In addition, and routinely, it is not possible to investigate airflow limitation in this age group. What is a fact is that it is the most common disease in childhood and causes a leading morbidity evidenced by school absenteeism, visits to emergency departments and hospitalizations [21]. Endotype is a subtype of condition defined by a different pathological mechanism. In asthma we have two endotypes; asthma Th-2 and asthma non-Th2 [22]. In the Th-2 endotype, the phenotype is basically eosinophilic and within it there is a subgroup with severe eosinophilic asthma refractory to conventional treatment [23]. The non-Th-2 endotype is also known as non-eosinophilic asthma (NEA). Chung classifies them as asthma "Th-2 high inflammation" and asthma "Th-2 low inflammation" (NEA) [24]. 50% of asthmatics will have the eosinophilic phenotype and the other 50% will have the NEA. There is not always a clear correlation between clinical phenotypes and molecular endotypes. In the Th-2 endotype, immunity is orchestrated by CD4+ T helper-2 that secrete interleukins IL-4, IL-5, IL-9, and IL-13, which lead to airway infiltration with eosinophils, mast cells, and B cells (which produce IgE in response to allergens) as described in the Th2 program [25]. The point is that these patients have peripheral eosinophilia and airway infiltration with eosinophils. Patients with NEA do not have significant eosinophilia and in this group, there are several phenotypes such as neutrophilic, paucigranulocytic and mixed pattern (eosinophils and neutrophils). These patients have various risk factors such as smoking, pollution, occupational exposure, recurrent infections, and obesity. It is difficult to separate NEA from COPD particularly in people of older age because they easily overlap, have similar spirometric patterns and similar molecular mechanisms that generate them. In both, the T-cell program is directed towards Th-1, Th-17, which attract neutrophils and macrophages but not eosinophils [26]. This type of asthma is more severe and responds less (like the mixed pattern) to corticosteroids. Although it is true that elevated IgE is a biomarker of Th2 asthma, early-onset, clearly allergic, it can also be found in late-onset non-allergic bronchial asthma. This happens, for example, in late-onset asthma exacerbated by staphylococcus aureus enterotoxins, where there is a specific IgE for enterotoxin (SE-IgE) and the biomarker predicts an evolution towards severe asthma, with frequent hospitalizations, use of oral steroids and deterioration of lung function. It seems that enterotoxin immunologically manipulates the airway mucosa, activating the Th2 program and generating increased IgE and tissue eosinophilia without mediating an allergic mechanism [27]. Allergic rhinitis affects up to 40% of the world population [28]. 40% of patients with allergic rhinitis have BA and 80-90% of asthmatics have allergic rhinitis. Patients with allergic rhinitis are 3 times more likely to develop BA than those who do not have it and 40- 60% of patients with BA have radiological data of chronic rhinosinusitis [29]. Chronic rhinosinusitis can occur with or without nasal polyposis. If it occurs with nasal polyposis, it is associated with more severe BA [30). The most common allergens in allergic respiratory disease today are, outdoor, tree pollen, grass pollen and weed pollen. From an internal environment, the most frequent are house dust mites, cats, dogs, and mold. Up to 30% of children and 2-10% of adults are impacted by allergic dermatitis and this entity is associated with BA, rhinitis, conjunctivitis, eosinophilic esophagitis, systemic immune activation, and food allergy [31]. Rarely food allergy is a trigger for asthma symptoms, but in patients with food-induced anaphylaxis, the co-existence of BA is a strong risk factor for more severe and even fatal reactions. Peanuts and nuts are the most commonly responsible foods [32]. Obviously, these associations have a strong allergic and immunological basis, reflecting type 2 immune inflammation. Quantification of peripheral eosinophils, sputum, and nasal exudate and specific and non-specific IgE attempts to document this diathesis, but markers of systemic inflammatory disease are not used because in BA there does not seem to be such activation.
Bronchopulmonary aspergillosis (ABPA) is a reaction of hypersensitivity to colonization of the airway by the ubiquitous mold aspergillus [33]. Affected patients typically have cough and exacerbations of asthma, and recurrent pulmonary infiltrates which can progress to bronchiectasis and pulmonary fibrosis [34]. Diagnostic criteria include a history of asthma or cystic fibrosis, elevated aspergillus specific IgE and IgG for aspergillus, increased serum IgE (> 1000 ng / ml or >410 iu / ml), wheal and-flare skin reaction for aspergillus antigen and high eosinophilia [35]. Exacerbations improve with the administration of prednisone.
At the gastrointestinal level, eosinophilia can be associated with eosinophilic gastroenteritis, eosinophilic colitis, and eosinophilic esophagitis. The latter condition is associated with allergic rhinitis, asthma, food allergy and atopy family history (20-80%) [36]. Findings of sensation of retrosternal burn, dysphagia and esophageal stenosis frequently confuse it with gastroesophageal reflux, so atopic diathesis and eosinophilia should suggest the diagnosis. Atopy seems to be an important factor but not mediated by IgE but by IgG4 which, as we know, can behave like a reagin. Th-2 cytokines promote eosinophilic infiltration and activation of TGF-β (transforming growth factor beta), remodeling and esophageal stenosis. Diet, esophageal dilations, and drugs are the therapeutic options. Budesonide (drinkable ampoules) 1mg / day swallowed in children, and 2-4 mg / day in adults, after meals, is an alternative. It works in Europe in the design of budesonide tablets.
Food allergy affects 6% of children and 2% of adults. Immune tolerance is an absence or decrease in a robust response of the immune system to autoantigens, that is, to pathogenic autoimmunity. It is possible that if this immune tolerance did not exist and the opposite occurred, uncontrolled immune hyperactivity, food allergy or autoimmune diseases would be much more prevalent [37]. Food allergy represents an adverse immune response to ingested proteins. The most common antigens ingested are cow's milk, eggs, peanuts, nuts, fish, sesame seeds, soybeans, mustards and sulphites (additives). Epitopes (antigenic determinants or fractions of macromolecules that are recognized by the immune system) are specific sequences to which antibodies bind, small size (20-70 Kd), and are water-soluble glycoproteins. They are usually resistant to acid or heat denaturation, and can remain intact after being processed, packed, cooked, or subjected to some type of chemical digestion [38]. Other antigens are denatured when cooked, for example eggs and milk in making cakes are generally well tolerated by allergic patients. Food allergy can give clinical manifestations in any system of the human economy, but skin (allergic dermatitis), gastrointestinal tract (gastroenteritis and eosinophilic esophagitis) and respiratory tract are the most frequently affected. Food allergy does not cause chronic respiratory symptoms and may be IgE-mediated or non-IgE-mediated (cell mediated). Non-allergic food events are metabolic disorders (eg, lactose intolerance), reactions to toxic pollutants (decomposing bacteria), reactions to pharmacologically active components (caffeine or tyramine generating migraine) [38]. In children allergic to peanuts, early, sequential, and progressive exposure, initiated between 4-11 months of age, reduces food allergy to peanuts by 86% and is maintained for a 60-month term. What happens is a progressive oral desensitization as an oral immune tolerance is created due to a shift in IgE that generated the allergy, an IgG4 that has a protective role since it inhibits the activation of basophils. This exposes the importance of immune tolerance as a therapeutic tool in food allergy [39].
Chronic urticaria is characterized by itchy welts, with central edema and surrounding erythema, and may be associated with cutaneous or mucosal angioedema. The acute form that lasts less than 6 weeks is usually allergic. Chronic urticaria (spontaneous or idiopathic) takes more than 6 weeks and is associated with elevated auto-IgE-mediated autoimmunity, and antithyroid antibodies and other autoimmune diseases (SLE). A third variant is induced by physical injury, cold, or by cholinergic pathway and can present with dermographism [40].
Parasitosis
A review of parasitosis in humans has recently been published in the journal [41]. The immune system of mammals uses the Th2 program to provide protection against parasites. In breve, IgE plays a primary pathogenic role in allergy, but has a protective role in defense against parasitic diseases such as helminths and protozoa. Many of these parasitosis have not only peripheral eosinophilia but also elevated serum IgE levels. As an example, Paragonimiasis is most prevalent in East Asia, West Africa, India, Central and South America, produced by Paragonimus westermani or Paragonimus sp. 50% of patients have marked eosinophilia and elevated levels of IgE [42] Tropical pulmonary eosinophilia (TPE) is a rare condition that occurs due to an unusual hypersensitivity reaction to filaria antigens of the parasites Wuchereria bancrofti and Brugia malayi [43]. Degenerated microfilariae release antigens that generate type I, II and IV immune responses, with high levels of IgE. Acute larval migration (Loeffler syndrome) is a self-limited pathology, caused by transpulmonary larval migration that occurs early in the normal life cycle of several helminths, including Ascaris lumbricoides, hookworm (Ancylostoma duodenale and Necator americanus) and Strongyloides stercolaris. Marked eosinophilia and high levels of IgE may appear and migratory infiltrates are seen on the chest radiograph [44]. Larva visceral migrans (acute toxocariasis) occurs when eggs of Toxocara canis or Toxocara catis are ingested, which are found in soils contaminated with dog or cat feces or by ingesting raw meat, particularly liver. The eggs generate larvae that penetrate the intestinal mucosa, enter the portal circulation and then into the systemic circulation with elevated IgE and eosinophilia [45]. Parasitosis not only stimulate the production of antiparasitic IgE antibody but can nonspecifically induce polyclonal IgE synthesis that results in highly elevated total serum IgE levels. Such polyclonal stimulation can diminish specific IgE antibody responses and cause saturation of mast cell Fc epsilon receptors, thus inhibiting allergic reactivity. This may represent a mechanism of immune evasion by the parasite. And on the other hand, the children with a strong atopic background demonstrated IgE responses concordant with an enhanced protective response against helminthic parasites and had significantly lower intensities of infection than their nonatopic counterparts. These observations support the concept that the atopic state has conferred a selective evolutionary advantage that could compensate for its involvement in allergic disease [46].
Human hyper-IgE syndromes (HIES)
HIES have a 50-year history of an association between high levels of IgE, and atopy, in a clinical context of bacterial and fungal infections in various organs, a reduced clinical and biological level of inflammation, and various non-hematopoietic developmental manifestations. All correspond to mutations. Job syndrome corresponds to autosomal dominant STAT 3 deficiency (AD STAT3 deficiency) (polymorphic mutation of the gene encoding the transcription factor). STAT 3 is a ubiquitous pleiotropic factor that involves responses from many cytokines including IL-6. STAT 3 is used together with JAK (janus kinase), as way to transmit signals of tissue development, homeostasis of the organism and defense of the host [47]. Clinically they generally have joint, skeletal, dental, and vascular abnormalities. Skin disorders include from neonatal rash to chronic atopic dermatitis. "Cold" skin and lung abscesses are usually caused by staphylococcus aureus (the similarity to the skin lesions of the prophet Job explains the name of the syndrome). Mucocutaneous candidiasis is common. At the immunological level, they present with high levels of IgE (>2.000 UI/ml), eosinophilia, memory B cell lymphopenia and low levels of Th17 [48]. DOCK 8 it is a deficiency of "dedicator of cytokinesis 8", a protein of lymphocytes. It is an autosomal recessive deficiency. (AR HIES). Its mutation leads to low levels of immune cells with eosinophilia, elevated IgE, lymphopenia of T cells and NK (natural killer), and susceptibility to viral and fungal infections. Shortly after birth they develop atopic dermatitis, allergic asthma, food allergy, and mucocutaneous candidiasis. Some authors consider that it qualifies more as a combined immunodeficiency (DIC) than as HIES. PGM 3 deficiency is a mutation of phosphoglucomutase 3. Its dysfunctionality affects downstream many fundamental processes of lipid and protein glycosylation. He has eosinophilia although not all patients have elevated IgE levels. They present with recurrent bacterial and viral infections, autoimmunity, severe allergic disease, developmental delay, and skeletal abnormalities [17]. AD-CARD deficiency leads to the dysfunctionality of CARD11, a normal white cell activating protein, with a clinical picture, similar previous one, and occurs with the deficiency of the IL-6 receptor, which, being mutated, does not allow IL-6 action. Many of these mutations have been recently described in the National Institute of Allergy and Infectious Diseases. Other syndromes with elevated IgE and immunodeficiency include: Wiskott-Aldrich, Di George, Omenn and Cornel-Netherton syndrome.
Neoplasms
Elevated IgE has been described in bronchogenic carcinoma, after bone marrow transplantation, and IgE myeloma. Sézary's syndrome (a peripheral T-cell neoplasm) has been associated with elevated IgE and/or eosinophilia when the malignant clone is of the CD4+ helper phenotype and produces an abnormal amount of the cytokine IL-4. Modestly elevated IgE has also been reported in B-cell chronic lymphocytic leukemia and in patients with Hodgkin's disease [49].
Other pathologies
HIV, RSV, leprosy, CMV, EBV and systemic candidiasis disease can present with elevated levels of IgE, but the finding does not always occur, and the elevation of immunoglobulin is not a diagnostic criterion, nor a biomarker of the disease or an objective therapeutic. The same happens in nephrotic syndrome, nephritis, liver disease, cystic fibrosis, Kawasaki disease, Guillain-Barré syndrome, and Kimura disease.
Biomarkers are characteristics that are objectively measurable and evaluable as indicators of a normal or pathological biological process or of a pharmacological response to therapeutic interventions. From this perspective, measuring total serum IgE is an adequate criterion for the diagnosis of ABPA, parasitosis or HIES, but in allergy, which constitutes its greatest practical utility, it has its limitations (Table 1). A normal total serum IgE value does not rule out allergic diathesis. This may require measuring specific IgE for one or more allergens. Furthermore, certain "allergic” pathologies can be mediated by IgG4 or IgG2 and present with normal IgE values. Unlike IgG and IgA, IgE has a lot of cross-reactivity, which makes it difficult to determine antigenic specificity. The cause of this variability is unknown, and this requires further investigation [50]. For example, tropomyosin Der p 10 from house dust mites is cross-reactive with cockroach and shrimp antigens, therefore it is not a relevant antigen for bronchial provocation tests [51]. The cat extract that tests positive for skin or the test positive for serum IgE is not relevant in bronchial provocation test. The first of the 2 allergens, Fel d2 (cat ovalbumin), is cross-reactive with pig albumin, giving pig-cat syndrome [52] and in alpha-gel syndrome, an antigen in beef cross-reacts with antigens from the saliva of ticks, chiggers and jellies that are used in marshmallow jellies and when appearing are not mediated neither by IgE nor by IgG4 but by IgM or IgG2. In the bronchial provocation’ test there are also unresolved needs and areas of uncertainty. Natural exposure to aeroallergens exposes patients to relatively large particle sizes with a broad spectrum of allergens and adjuvants. In the laboratory test the sizes are smaller and variable. In natural exposure, local inflammation leads to prolonged bronchial hyperresponsiveness. In the bronchial provocation test, hyperresponsiveness is transient, which implies that many exacerbations may not necessarily be due to natural exposure to a given antigen (which gave a positive test) but due to exposure to a viral trigger. Skin tests are generally not convincing in defining the importance of sensitivity. Specific IgE or IgG subunits are required to be measured. Total serum IgE has a limited value. There are a series of mitogenic factors, not necessarily allergenic, that can stimulate its production, such as viruses, bacteria, helminths, and environmental pollution (diesel, cigarette smoke). As always in medicine, a good history can determine the relevance or not of a laboratory finding [7]. The 3 most frequently used methods to measure specific IgE are: RAST (radioallergosorbent test) and derivates, FAST (fluorescentallergosorbent test) and ELISA (enzyme-linked immunosorbent assay).
Table 1. The normal value of IgE in KU/L are shown according to age
IgE-mediated autoimmunity (autoallergy) involves IgE as an antibody against the patient's own antigens (autoantigens). To involve this in the mechanism of immunological pathogenesis, it is necessary to identify IgE as an autoantibody and define the target autoantigens. IgE's primary role is host defense. This was the primary function that appeared during the evolutionary development of the antibody to eliminate worm-like parasites (helminths) and protozoa and environmental substances such as toxins, poisons, and xenobiotics. The pathogenic role of IgE appears with the acquisition of an antigenic power of environmental proteins. The recognition of self-antigens by IgE is an unclear phenomenon and the role of autoimmunity by IgE in autoimmune diseases is poorly understood [53]. IgE appears to be able to recognize the patient's own molecules as autoantigens that have undergone phosphorylation. In bullous pemphigus, IgE and IgG have been described, recognizing phosphorylated epitopes. What is not clear if there is a preferential recognition for IgE or they can be recognized as autoantigens by the entire immune system [54]. Methods for detecting autoreactive IgE have high variability and low reproducibility and this is an area that requires further research to improve detection methods.
Self-reactive IgE has been detected in several entities. In atopic dermatitis with a variable frequency (23-91%), particularly in severe cases and directed against keratinocytes and nuclear antigens [55]. Bullous pemphigoid is a blistering skin disease that affects older patients. While it is true, they have auto-IgA and auto-IgG against hemidesmosomal proteins, they also have auto-IgE against antigenic proteins (BP180, type XVII collagen and intracellular BP-230). BPs are adhesion molecules to the cellular substrate expressed in keratinocytes. IgG and IgE autoantibodies share the same epitope. Autoantibodies promote cell degranulation and auto-IgE deposition on the basement membrane of the skin [56]. Pemphigus vulgaris and pemphigus foliaceus are bullous autoimmune skin diseases associated with auto-IgG directed against desmoglin-3 (desmosomal adhesion molecule), but epidermal auto-igE deposits directed against the same antigen have also been described. Chronic idiopathic urticaria is not generally considered a classic autoimmune disease, but a subgroup of patients has auto-IgG and auto-IgE against FCϵRI, against dsDNA, against thyroglobulin and thyroperoxidase, and present with thyroiditis and SLE. Anti-IL-24 IgE correlates with activity’s disease. 32% of patients with SLE have auto-IgE against dsDNA and this finding correlates with disease activity, but not with the presence of atopic diseases. A controversial point is whether they represent autoimmunity per se or are the product of a cross-reaction against allergens. IgE anti-dsDNA appears to be associated with lupus nephritis [57]. In rheumatoid arthritis, auto-IgE directed against nuclear antigens and citrullinated protein have been described, particularly in patients with neutropenia. The phenomenon has also been described in autoimmune uveitis, multiple sclerosis, Graves' disease, and autoimmune pancreatitis. There is little or no evidence in detail in bronchial asthma, allergic rhino-conjunctivitis, and chronic rhinosinusitis with nasal polyposis [50,56].
IgE-mediated chronic diseases are treated with antihistamines, corticosteroids, other anti-inflammatory drugs, and biologics. Understanding the Th2 program has made it possible to design recombinant humanized monoclonal antibodies that block Th2 cytokines, or their receptors, modifying the downstream signals of effector cells and reducing the release of pro-inflammatory mediators or chemotaxis from inflammatory cells. Omalizumab (OmAb) is a monoclonal antibody that acts by several mechanisms. Sequesters free serum IgE, accelerates dissociation of mIgE (membrane receptor-bound-IgE) from the receptors, and reduces blood, sputum, and bronchial biopsy eosinophilia in bronchial asthma. This is because OmAb induces eosinophilic apoptosis. Patients treated with OmAb have elevated levels of anexin, a marker of eosinophilic apoptosis [58]. OmAb was designed to bind IgE at the Fc fragment, at the Cϵ3 locus, in the same domain where IgE binds to the FcϵRI and FcϵRII receptors. The basic idea is to sequester IgE and induce eosinophilic apoptosis and reduce allergic inflammation [59]. A proposed mechanism, although not validated, is that OmAb can bind to antigens, behaving with a competitive inhibitor [60]. It is administered subcutaneously and is slowly absorbed. The half-life is 26 days. It is usually dispensed once a month, reaching a maximum concentration peak between the 7-8th day and the OmAb-IgE complex is eliminated by the reticuloendothelial system. The dose depends on anthropometric variables and IgE levels, but usually ranges between 150-300 mg SC every 4 weeks (Figure 3). Continuous treatment with OmAb marginally increases the risk of arterial thrombosis and has a negative impact on cardiac output and cerebral circulation. Side effects include erythema and pain at the injection site up to systemic anaphylaxis.
Figure 3. Monoclonal antibodies in bronchial asthma
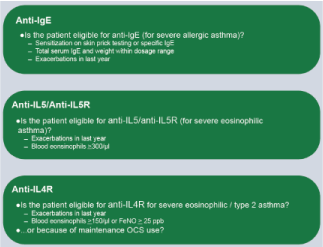
The indications for each group of monoclonal antibodies are listed.
10% of chronic asthmatic patients respond poorly to conventional treatment. In the group of persistent and moderate to severe asthmatics with a clear allergic component (Th2 endotype) in which control is not achieved, the use of OmAb is indicated, from 6 years of age. Initial studies already demonstrated a reduction of up to 50% of IGCs (inhaled corticosteroids) and even their discontinuation [61]. More recent works add a reduction in symptoms and exacerbations and limits remodeling. [62]. The standard duration of treatment with OmAb has not been established to date. A follow-up study showed that, after six years of OmAb treatment, most patients had mild and stable asthma in the ensuing three years after treatment discontinuation [63]. It has been suggested that the persistence of the effects of OmAb may be due to its ability to curtail airway remodeling in patients with asthma.
The use of OmAb in urticaria has focused mainly on CSU with autoimmune form. Particularly, in those older than 12 years and who persist symptomatic [64]. The effect of OmAb on CSU with or without angioedema has been demonstrated in several double-blinded randomized placebo-controlled studies [65]. Although the OmAb dose for CSU is set at 300 mg every 4 weeks, a dose of 150 mg every 4 weeks also achieves an effect in some patients, and in other cases the dose needs to be increased to 300 mg every two weeks. Nevertheless, it is still unclear how OmAb works in CSU: in addition, the fact that OmAb is not effective in all patients suggests the involvement of mechanisms/pathways in CSU other than the IgE cascade.
Systemic mastocytosis is a heterogeneous disorder that results from abnormal proliferation and accumulation of mast cells in more than one organ. OmAb reduces the expression, in these cases, of FcϵRI in circulating basophils and mast cells, but it does not work in all patients. In HIES, clinical improvement has been described in some patients. In eosinophilic gastroenteritis, the use of OmAb reduces peripheral eosinophils and antral and duodenal eosinophils by 35-45%. It effectively blocks CD23-mediated allergen binding to B-cells [66].
There is a close relationship between asthma and allergic rhinitis. For this reason, OmAb was expected to be effective in the treatment of concomitant rhinitis in patients with asthma. Indeed, in one trial, the odds ratio for a positive effect on rhinitis was 3.56, indicating that the probability of improvement was three and a half times higher in subjects treated with OmAb [67]. In nasal polyposis, the results of using OmAb are less obvious since this condition appears in nonallergic patients. Omalizumab has also demonstrated its clinical relevance in patients with allergic bronchopulmonary aspergillosis (ABPA) with or without CF [68].
In allergic dermatitis and bullous pemphigus, the results are controversial. In food allergies, the use of OmAb in conjunction with oral desensitization has therapeutic potential. In atopic keratoconjunctivitis, which is a severe hypersensitivity reaction that can cause loss of visual acuity and blindness, benefit has been reported [69].
B cells have an extra 52 amino acid fragment called Cϵmx that is located between the Cϵ4 domain of IgE and its membrane anchoring segment. Quilizumab is a monoclonal antibody that targets this segment, inducing apoptosis of IgE + cells, reducing free IgE levels, however it does not produce significant clinical benefits in allergic asthma. Specific antibodies against IgE such as IgE-CD3 that reduce free serum IgE and levels of IgE + B cells, as well as other prototypes that reduce these cells and the number of plasma cells that generate IgE is being investigated, but so far only OmAB is approved by FDA as a monoclonal antibody that directly aims to reduce IgE in allergic asthma and chronic urticaria [70].
Other monoclonal antibodies that impact other Th2-cytokines are used, particularly in severe allergic asthma. Mepolizumab, reslizumab, and benralizumab are antibodies that target IL-5 or its receptor; basic interleukin in the attraction and activation of eosinophils [71]. IL-4 and IL-13 (which activate the synthesis of IgE) act by stimulating the α-subunit of the IL-4 receptor. Dupilumab inhibits this receptor and therefore the actions of both interleukins, impacting the synthesis of IgE, but not directly the serum IgE [72,73] (Figure 3).
IgE plays a primary role in defense against helminths and protozoa, but also in the pathogenesis of allergic diseases in mammals.
IgE not only plays a preponderant role in the diagnosis of many diseases allergic to external antigens, but also as an antibody directed against autoantigens (autoimmunity).
The detection of autoreactive IgE requires future research to improve its variability and reproducibility and to define more and better its role in autoimmune diseases.
The development of monoclonal antibodies anti-IgE or directed against Th2 cytokines has put IgE immunoglobulin back on the research table.
Several new molecules directed against the cascade of synthesis or molecular effects or signals of the antibody are on the research radar, and it is possible that their clinical use will impact the natural history of IgE-related diseases in the future.
This work was only carried out by the author. Author AA contributed on the planning, data collection, data analysis, writing and critical review. AA read and approved the final manuscript.
None.
None.
- Foster S (2011) How can you help people with allergies? Pharm J 286: 535‐ 538.
- Hellman LT, Akula S, Thorpe M, Fu Z (2017) Tracing the origins of IgE, mast cells, and allergies by studies of wild animals. Front Immunol 8: 1749. [Crossref]
- Vernersson M, Aveskogh M, Hellman L (2004) Cloning of IgE from the echidna (Tachyglossus aculeatus) and a comparative analysis of ε chains from all three extant mammalian lineages. Dev Comp Immunol 28: 61‐75. [Crossref]
- Akula S, Mohammadamin S, Hellman L (2014) Fc receptors for immunoglobulins and their appearance during vertebrate evolution. PLoS One 9: e96903. [Crossref]
- Pritchard DI, Falcone FH, Mitchell PD (2020) The evolution of IgE-mediated type I hypersensitivity and its immunological value. Allergy 00: 1-17.
- Ishizaka K, Ishizaka T, Hornbrook M (1966) Physicochemical properties of reaginic antibody. V. Correlation of reaginic activity with gamma-E-globulin antibody. J Immunol 97: 840-853. [Crossref]
- Platts-Mills TAE, Schuyler AJ, Erwin EA, Commins SP, Woodfolk JA (2016) IgE in the diagnosis and treatment of allergic disease. J Allergy Clin Immunol 137: 1662-1669. [Crossref]
- Hamilton RG, MacGlashan DW Jr, Saini SS (2010) IgE antibody-specific activity in human allergic disease. Immunol Res 47: 273-284. [Crossref]
- Balbino B, Conde E, Marichal T, Start IP, Reber LL (2018) Approaches to target IgE antibodies in allergic disease. Pharmacol Ther 191: 50-64. [Crossref]
- Sutton BJ, Davies AM (2015) Structure and dynamic of IgE-receptor interactions: Fc epsilon RI and CD23/Fc epsilon RII. Immunologic Rev 268: 222-235. [Crossref]
- Wu LC, Sarrin AA (2014) The production and regulation of IgE by the immune system. Nat Rev Immunol 14: 247-259. [Crossref]
- Dhaliwal B, Pang MO, Keeble AH, James LK, Gould HJ, et al (2017) IgE binds asymmetrically to its B cell receptor CD23. Scient Rep 7: 45533.
- Gould HJ, Beavil R (1998) Immunoglobulin E. In: Encyclopedia of Immunology. (2 Edn).
- Alvarado A (2019) Differences, similarities and controversies between bronchial asthma and chronic obstructive pulmonary disease. Clin Res Trials 5: 1-12.
- Yao Y, Chen CL, Yu D, Liu Z (2020) Roles of follicular helper and regulatory T cells in allergic disease and allergen immunotherapy. Allergy.
- Stokes J (2017) Anti-IgE treatment for disorders other than asthma. Front Med (Lausanne) 4: 152. [Crossref ]
- Zhang Q, Boisson B, Béziat U, Puel A, Casanova JL (2018) Human hyper-IgE syndrome: singular or plural? Mamm Genome 29: 603-607. [Crossref]
- Wernersson S, Pejler G (2014) Mast cell secretory granules: Armed for battle. Nat Rev Immunol 3: 681-686. [Crossref]
- Rosenberg HF, Dyer KD, Foster PS (2013) Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. 13:9-22. [Crossref]
- To T, Stanojevic S, Moores G, Gerson AS, Baterman ED, et al (2012) Global asthma prevalence in adults: finding from the cross-sectional World Health Survey. NMC Public Health 12: 204. [Crossref]
- GINA (2020) Global Strategy for Asthma Management and Prevention. Global Initiative for Asthma.
- Esteban-Gorgojo I, Antolin-Amérigo D, DomÃnquez Ortega J, Quirce S (2018) Non-eosinophilic asthma: current perspectives. J Asthma Allergy 11: 267-281. [Crossref]
- Ortega HG, Lin LM, Pavord ID, Bruselle GG, FitzGerald JM, et al (2014) Mepolizumab treatment in severe eosinophilic asthma. N Engl J Med 371: 1198-1207. [Crossref]
- Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, et al (2014) International ERS/ATS guidelines on definition, evaluation, and treatment of severe asthma. Eur Respir J 43: 343-373. [Crossref]
- Barnes PJ (2017) Cellular and molecular mechanisms of asthma and COPD. Clin Sci (Lond) 131: 1541-1558. [Crossref]
- Postma DS, Rabe KF (2015) The Asthma-COPD overlap syndrome. N Engl J Med 373: 1241-1249. [Crossref]
- Bachert C, Humbert M, Hannia NA, Zhang N, Holgate E, et al (2020) Staphylococcus aureus and its IgE-inducing enterotoxins in asthma: current knowledge. Eur Respir J 55: 1901592. [Crossref]
- Tong MCF, Lin JSC (2015) Epidemiology of allergic rhinitis throughout the world. In: Akdis CA, Hellings PW, Agache I (Eds) Global Atlas of Allergic Rhinitis and Chronic Rhinosinusitis. Zurich, Zwitzerland. European Academy of Allergy and Clinical Immunology pp: 62-63. [
- Schmitt J, Stadler E, Küster D, Wüstenberger EC (2016) Medical care and treatment of allergic rhinitis: a population-based cohort study based on routine health care utilization data. Allergy 71: 850-858. [Crossref]
- Hamilos DL (2011) Chronic rhinosinusitis. Epidemiology and medical management. Allergy Clin Immunol 128: 693-707. [Crossref]
- Lyons JJ (2015) Atopic dermatitis in children: clinical features, pathophysiology, and treatment. Immunol Allergy Clin North Am 35: 161-183. [Crossref]
- Bock SA, Munoz-Furlong A, Sampson HA (2007) Further fatalities caused by anaphylactic reactions to food, “2001-2006”. J Allergy Clin Immunol 119: 1016-1018. [Crossref]
- Cottin V, Cordier JF (2012) Eosinophilic lung diseases. Immunol Allergy Clin North Am 32: 557-586. [Crossref]
- Agarwal R, Chakrabarti A, Shah A, Gupta D, Meis JF, et al (2013) Allergic bronchopulmonary aspergillosis: review of literature and proposal a new diagnostic and classification criteria. Clin Exp Allergy 43: 850-873. [Crossref]
- Pinal-Fernández I, Selva-O´Callaghan A, Grau JM (2014) Diagnosis and classification of eosinophilic fasciitis. Autoimmune Rev 13: 379-382. [Crossref]
- Reed CC, Dellon ES (2019) Eosinophilic esophagitis. Med Clin North Am 103: 29-42. [Crossref]
- Bluestone JA, Anderson M (2020) Tolerance in the age of immunotherapy. N Engl J Med 38: 1156-1166. [Crossref]
- Waserman S, Bégin P, Watson W (2018) IgE-mediated food allergy. Allergy Asthma Clin Immunol 14: 55. [Crossref]
- Du Toit G, Roberts G, Sayre PH, Bahnson HT, Radulovic S, et al (2015) Randomized trial of peanut consumption in peanut allergy. N Engl J Med 372: 803-813. [Crossref]
- Navinés-Ferrer A, Serrano-Candelas E, Molina-Molina GJ, Martin M (2016) IgE-related chronic diseases and Anti-IgE-based treatments. J Immunol Res 2016: 1-12. [Crossref]
- Alvarado A (2020) The Eosinophil: Physiology and Pathology. Clin Res Trials 6: 1-11.
- Fürst T, Keiser J, Utzinger J (2012) Global burden of human food-borne trematodiasis: a systemic review and metanalysis. Lancet Infect Dis 12: 210-221. [Crossref]
- Vijayan VK (2007) Tropical pulmonary eosinophilia: pathogenesis, diagnosis, and management. Curr Opin Pulm Med 13: 428-433. [Crossref]
- Jourdan PM, Lamberton PHL, Fenwick A, Addiss DG (2018) Soil-transmitted helminth infections. Lancet 391: 252-265. [Crossref]
- Smith H, Holland C, Taylor M, Magnaval JF, Schantz P, et al (2009) How common is human toxocariasis? Toward standardizing our knowledge. Trends Parasitol 25: 182-188. [Crossref]
- Lynch NR, Hagel IA, Palenque, Di Prisco MC, Escudero JE, et al (1998) Relationship between helminthic infection and IgE response in atopic and nonatopic children in a tropical environment. J Allergy Clin Immunol 101: 217-221. [Crossref]
- O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, et al (2015) The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Ann Rev Med 66: 311-328. [Crossref]
- Schezawinska-Poplonyk A, Kyeler Z, Püetrwcha B (2011) The hyperimmunoglobulin E syndrome: clinical manifestations diversity in primary immune deficiency. Orphanet J Rare Dis 6: 76. [Crossref]
- Ellis AK, Wasermann S (2009) Hodgkin´s lymphoma presenting with markedly elevated IgE: a case report. All Asth Clin Immun 5: 12. [Crossref]
- Maurer M, Altrichter S, Schmetzer O, Schoffel J, Church MK (2018) Immunoglobulin E-mediated autoimmunity. Front Immunol 9: 1-17. [Crossref]
- Bronnert M, Mancini J, Brinbaum J, Agabriel C, Labeuf V, et al (2012) Component-resolved diagnosis with commercially available D. pteronyssinus Der p1, Der p2, and Der p10: relevant markers of house mite allergy. Clin Exp Allergy 42: 1406-1415. [Crossref]
- Posthumus J, James HR, Lanc CJ, Matos LA, Platts-Mills TA, et al (2013) Initial description of pork-cat syndrome in the United States. J Allergy Clin Immunol 131: 923-925. [Crossref]
- Panaszek B, Pawlowicz R, Grzegrzolka J, Obojski A (2017) Autoreactive IgE in chronic spontaneous/idiopathic urticaria and basophil/mastocyte priming phenomenon, as a feature of autoimmune nature of the syndrome. Arch Immunol Ther Exp (Warsz) 65: 137-143. [Crossref]
- van Beek N, Schulze FS, Zillikens D, Schmidt E (2016) IgE-mediated mechanisms in bullous pemphigoid and other autoimmune bullous diseases. Expert Rev Clin Immunol 12: 267-277. [Crossref]
- Tang TS, Bieber T, Williams HC. (2012) Does “autoreactivity” play a role in atopic dermatitis? J Allergy Clin Immunol 129: 1209-1215. [Crossref]
- Sanjuan MA, Sagar D, Kolbe K (2016) Role of IgE in autoimmunity. J Allergy Clin Immunol 137: 1651-1661. [Crossref]
- Hanaoka H, Okazaki Y, Satoh T, Kaneko Y, Yasuoka H, et al (2012) Circulating anti-double-stranded DNA antibody-secreting cells in patients with systemic lupus erythematosus: a novel biomarker for disease activity. Lupus 21: 1284-1293. [Crossref]
- Massanari M, Holgate ST, Busse WW, Jimenez P, Kianifard F, et al (2010) Effect of omalizumab on peripheral blood eosinophilia in allergic asthma. Respir Med 104: 188-196. [Crossref]
- Babu KS, Polosa R, Morjaria JB (2013) Anti-IgE—emerging opportunities for Omalizumab. Exp Opin Biolog Ther 13: 765–777. [Crossref]
- Chang TW (2000) The pharmacological basis of anti-IgE therapy. Nat Biotechnol 18: 157-162. [Crossref]
- Walker S, Monteil M, Phelan K, Lasserson tJ, Walters EH (2006) Anti-IgE for chronic asthma in adults and children. Cochrane Database Syst Rev 2: CD003559. [Crossref]
- Mauro M, Incorvaia C, Formigoni C, Elia R, Russello M, et al (2012) The anti-IgE antibody omalizumab as a probe to investigate the role of IgE in pathology. Panminerva Med 54: 305-312. [Crossref]
- Nopp A, Johansson SGO, Adédoyin J, Ankerst J, M. Palmqvist M, et al (2010) After 6 years with Xolair; a 3-year withdrawal follow-up. Allergy: Eur J Allergy Clin Immunol 65: 56-60. [Crossref]
- Kaplan AP (2014) Therapy of chronic urticaria: a simple, modern approach. Ann All Asth Immunol 112: 419-425. [Crossref]
- Kaplan A, Ledford D, Ashby M, Canvin J, Zazzali JL, et al (2013) Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol 132: 101-109. [Crossref]
- Stone KD, Prussin C (2008) Immunomodulatory therapy of eosinophil-associated gastrointestinal diseases. Clin Exp Allergy 38: 1858–1865. [Crossref]
- Humbert M, Boulet LP, Niven RM, Panahloo Z, Blogg M, et al (2009) Omalizumab therapy: patients who achieve greatest benefit for their asthma experience greatest benefit for rhinitis. Allergy 64: 81-84. [Crossref]
- Lehmann S, Pfannenstiel C, Friedrichs F, Kröger K, Wagner N (2014) Omalizumab: a new treatment option for allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Ther Adv Respir Dis 8: 141-149. [Crossref]
- Williams PB, Sheppard Jr JD (2005) Omalizumab: a future innovation for treatment of severe ocular allergy? Exp Opin Biol Ther 5: 1603–1609. [Crossref]
- Hu J, Chen J, Ye L, Cai Z, Sun J (2018) Anti-IgE therapy for diseases: from neutralizing IgE antibodies to eliminating IgE+ B cells. Clin Transl Allergy 8: 27. [Crossref]
- Sullivan A, Ward C, Casey D, Flynn D, Hunt EB (2020) Anti-IL-5 therapy and the microbiome in asthma. Eur Respir J 56: 5279.
- Maspero JF, Halpin DMG, Jackson D, Harania NA, Castro M (2020) Effect of dupilumab on oral corticosteroid use in severe asthma patients with improving lung function. Eur Respir J 56: 5282.
- Wechsler ME, Ford LB, Maspero JF, Pavord ID, Langton D, et al (2020) Late breaking abstract. Dupilumab long-term safety and efficacy in patients with asthma: LIBERTY and ASTHMA TRAVERSE. Eur Respir J 56: 4613.