IgM nephropathy (IgMN) is a rare disease presenting as idiopathic nephrotic syndrome (INS) and acute renal failure both in children and adults. The disease is defined by its immunohistologic features, the presence of immunoglobulin M (IgM) in the mesangial regions of the glomeruli in a diffuse (all glomeruli) and global (the entire glomerulus) pattern [1-7].
26-year-old Caucasian male presented with complaints of progressive edema of the legs. He complained of a four-week history of progressive edema of the legs, periorbital edema, and 15-pound weight gain during that period. He also noticed frothy looking urine. He denied any fever, chills, nausea, vomiting, diarrhea, URTI symptoms, joint pains, rashes. No significant past medical, surgical history. No family history of kidney disease or ESRD. On vital signs, his blood pressure was 110/80 mm Hg. On physical exam, he had periorbital edema and 1+ pedal edema in bilateral lower extremities. Serum creatinine was 1.2 mg/dl. Urine analysis showed significant proteinuria and hematuria with RBC casts. 24-hour urine showed 4.3 grams of protein. Cholesterol 350 mg/dl, triglycerides 550 mg/dl. C3 and C4 levels were within normal limits. ANA, p-ANCA, and c-ANCA were negative. He underwent an ultrasound-guided kidney biopsy for diagnosis, and the pathology revealed IgM glomerulonephritis. Pt was initiated on steroids with no evidence of improvement in 2 months and no change in creatinine value. Since the patient was steroid-resistant, the plan was to start on immunosuppressive medications. He was started on cyclosporine. However, he developed acute renal failure with an elevation of creatinine to 2.7 mg/dl after four weeks of treatment which was then stopped and creatinine improved to 1.5 md/dl. Other options of immunosuppressive medications were discussed, he refused to take cyclophosphamide, so mycophenolate was started at 360 mg twice daily. Over a period of 6 months, proteinuria decreased to 0.5 mg/dl in 24 hours and creatinine stabilized at 1.3 gm/dl with a resolution of symptoms. Mycophenolate has been continued for one more year, his numbers have been stable on follow up for two years until the present time.
IgM glomerulonephritis presents as a nephrotic syndrome in adults and children. The frequency of IgMN reported in studies has varied widely from 2% to 18.5% [1,5-9]. Etiology is unknown for the development of the primary form of IgMN. However, some systemic diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis, diabetes mellitus, paraproteinemia, and Alport’s syndrome [10-15], showed deposits of IgM in the glomeruli.
Disease pathogenesis is by classical immune complex-mediated activation of the complementary cascade, triggered by antigens in the food and environment resulting in glomerular mesangium injury [3], immune complex formation and deposition of C1q, C3 and C4 deposits along with IgM in the mesangium. Abnormalities of T-lymphocyte regulatory role or a disturbance in the mesangial cell clearance of the passively trapped immune complexes have also been hypothesized [3].
Diagnosis depends on the pathologic evaluation of renal biopsy by LM, IF, and EM. The pathologic findings need to be correlated with clinical and serological studies to exclude the secondary causes of IgMN.
LM findings in IgMN can vary from minor changes to variable degrees of mesangial proliferation, usually of mild to moderate degree, to focal segmental glomerulosclerosis (FSGS) pattern accompanied by adhesion formation with the Bowman’s capsule [3-5]. Tubular atrophy and interstitial scarring are also commonly observed on renal biopsies of IgMN at the time of diagnosis and are usually mild [4,5,9,10] (Figure 1).
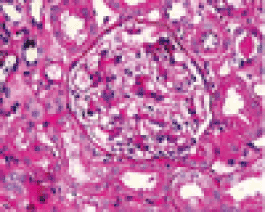
Figure 1. Renal biopsies of IgMN.
It is difficult to distinguish IgMN from minimal change disease (MCD) on LM examination alone and requires IF and EM to confirm the diagnosis.
IgMN is diagnosed based on the immunofluorescence microscopy which shows diffuse and global mesangial positivity of IgM of at least 1+intensity on a scale of 0-3+ (where 0 is absent, 1+ is mild, 2+ moderate, and 3+ is marked), deposits of IgA and IgG were also noted [4,5]. C3 and C1q are also noted in a good percentage of biopsies with IgM deposits [4,5,11] (Figure 2).
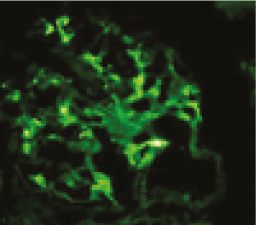
Figure 2. Biopsies with IgM deposits.
EM is not of much significance for diagnosis of IgMN. EM shows electron-dense deposits in the mesangium and para mesangium, along with variable degrees of mesangial cell proliferation, matrix expansion and fusion of foot processes [2,3].
IgMN occurs predominantly in children and young adults, but it can occur at any age. It is more common in males with an early mean age of onset [9]. The most common presentation is idiopathic NS with hematuria (HU) or asymptomatic urinary abnormalities (AUA) [2-5]. Hypertension has also been noted in some patients as presenting feature [2,3,5]. And its prevalence increases with duration of the disease approaching 50% at 15 years of follow-up [4].
Isolated HU or proteinuria-hematuria (PU-HU) are biopsy indications [4].
Renal failure was noted to be 35% at 15 years of follow-up, and ESRD developed in 23% of cases in one study [4].
Bad prognostic indicators were hypertension, proteinuria on initial presentation, FSGS, tubular atrophy and interstitial fibrosis from the features noted on biopsies [9].
Corticosteroids are the mainstay of treatment. The steroid response pattern in IgMN varies, and it was also found in about one-third of the children with INS5. Immunosuppressive agents have not been used much in the treatment of IgMN unless patient exhibits steroid resistant pattern. Oral cyclophosphamide has been used with response rates of up to 50% [4,8]. Mycophenolate has been used in one study for treatment in steroid dependent and resistant nephrotic syndrome with good results [12]. There is no adequate data or studies that were done on other immunosuppressive medications in IgMN. Rituximab has also been used in the treatment if there is no improvement or toxicity develops with immunosuppressive medications [12]. Disease recurrence after renal transplantation can be treated with anti-CD antibodies (rituximab), in combination with plasma exchanges, and immunoglobulins [13-16].
This is an unusual case of IgM glomerulonephritis that was successfully treated with steroids and mycophenolate combination achieving remission.
IgMN is a rare and less frequently reported cause of renal morbidity in both children and adults. It shows a spectrum of pathological changes ranging from minor changes to FSGS. Immunofluorescence is the cornerstone for its diagnosis and corticosteroids are the first line therapy. IgMN could be steroid-resistant, and it is important to distinguish it from MCD based on the steroid response and other pathological features. Adult females have a good prognosis, compared with patients with NS or proteinuria (PU) [4].
- Cohen AH, Border WA, Glassock RJ (1978) Nehprotic syndrome with glomerular mesangial IgM deposits. Lab Invest 38: 610-619. [Crossref]
- Lawler W, Williams G, Tarpey P, Mallick NP (1980) IgM associated primary diffuse mesangial proliferative glomerulonephritis. J Clin Pathol 33: 1029-1038.
- Hsu HC, Chen WY, Lin GJ, Chen L, Kao SL, et al. (1984) Clinical and immunopathologic study of mesangial IgM nephropathy: report of 41 cases. Histopathology 8: 435-446. [Crossref]
- Myllymäki J, Saha H, Mustonen J, Helin H, Pasternack A (2003) IgM nephropathy: clinical picture and long-term prognosis. Am J Kidney Dis 41: 343-350. [Crossref]
- Mubarak M, Kazi JI, Shakeel S, Lanewala A, Hashmi S, et al. (2011) Clinicopathologic characteristics and steroid response of IgM nephropathy in children presenting with idiopathic nephrotic syndrome. APMIS 119: 180-186. [Crossref]
- Mubarak M, Kazi JI (2012) IgM nephropathy revisited. Nephrourol Mon 4: 603-608. [Crossref]
- Bhasin HK, Abuelo JG, Nayak R, Esparza AR (1978) Mesangial proliferative glomerulonephritis. Lab Invest 39: 21-29. [Crossref]
- Hamed RM (2003) Clinical significance and long-term evolution of mesangial proliferative IgM nephropathy among Jordanian children. Ann Saudi Med 23: 323-327. [Crossref]
2021 Copyright OAT. All rights reserv
- Singhai AM, Vanikar AV, Goplani KR, Kanodia KV, Patel RD, et al. (2011) Immunoglobulin M nephropathy nephropathy in adults and adolescents in India: a single-center study of natural history. Indian J Pathol Microbiol 54: 3-6. [Crossref]
- Mokhtar GA (2011) IgM nephropathy: clinical picture and pathological findings in 36 patients. Saudi J Kidney Dis Transpl 22: 969-975.
- Zeis PM, Kavazarakis E, Nakopoulou L, Moustaki M, Messaritaki A, et al. (2001) Glomerulopathy with mesangial IgM deposits: long-term follow up of 64 children. Pediatr Int 43: 287-292. [Crossref]
- Tasanarong A, Thitiarchakul S. Mycophenolate mofetil treatment in primary glomerular disease: one-year follow-up in steroid dependent and resistant nephrotic syndrome. J Med Assoc Thai 89: S218-S227.
- Betjes MG, Roodnat JI (2009) Resolution of IgM nephropathy after rituximab treatment. Am J Kidney Dis 53: 1059-1062. [Crossref]
- Gu J, Xia Y, Mao J, Fu H, Liu A (2012) Rituximab followed by mycophenolate mofetil in children with IgM nephropathy. Indian Pediatr 49: 831-833. [Crossref]
- Salmon AH, Kamel D, Mathieson PW (2004) Recurrence of IgM nephropathy in a renal allograft. Nephrol Dial Transplant 19: 2650-2652. [Crossref]
- Westphal S, Hansson S, Mjornstedt L, Molne J, Swerkersson S, et al. (2006) Early recurrence of nephrotic syndrome (immunoglobulin m nephropathy) after renal transplantation successfully treated with combinations of plasma exchanges, immunoglobulin, and rituximab. Transplant Proc 38: 2659-2660. [Crossref]