Abstract
The technique of fluorescence microscopy has become an essential tool in the biomedical sciences. In all living cells, cellular, physiological, and pathological phenomena are accompanied by ionic changes. Then, the development of this technique that allows the measurement of intracellular ion activities has contributed substantially to understanding of normal and abnormal cellular function. In addition to determining the intracellular ion concentration, this technique has allowed the spatial analysis of ion activity at the subcellular level. Calcium ion (Ca2+) is a basic requirement cellular function. This ion participates of the regulation of several physiological cellular functions in all living cells. However, the dysregulation of its intracellular homeostasis can trigger pathological process, including the cell injury and death. The fluorescence microscopy techniques using fluorescent Ca2+ indicators have been developed to precisely analyze intracellular Ca2+ signaling in living cells. We have used this technique to study the precise role of the intracellular Ca2+ signaling in different physiological and pathological process, including in cardiovascular diseases. These studies can contribute decisively for understanding of the role of intracellular Ca2+ signaling in the pathophysiology of hypertension, and development of more efficient, and safer, pharmacological antihypertensive strategies to treat human hypertension.
Key words
Fluorescence microscopy, Fluorescent Ca2+ indicators, Intracellular Ca2+ signalling
Introduction
Ca2+ is a basic requirement for cellular function. This ion acts as a universal second messenger in a variety of living cells. The beginning of life, the act of fertilization, is regulated by Ca2+ [1]. Numerous functions of all types of cells are regulated by Ca2+, including contraction, neurotransmitter/hormone secretion, and cell proliferation and differentiation [1]. More than a century ago, Ringer demonstrated that Ca2+-containing perfusate was required for the contraction of frog heart [2]. In the 1940s, it was shown that an injection of Ca2+ into a muscle fiber induced contraction, but K+ or Na+ did not [3]. In the 1960s, it was shown that the sarcoplasmic reticulum is an important intracellular Ca2+ storage site in muscle fibers [4].
In living cells, the fine control intracellular Ca2+ concentration ([Ca2+]i) involves several Ca2+ regulatory mechanisms, including Ca2+ channels, Ca2+ exchangers, and Ca2+ pumps [1]. Since the 1920s, researchers have attempted to measure [Ca2+]i, but few were successful. In the 1960´s, Ridgway and Ashley introduced the first reliable measurements of [Ca2+]i by injecting the photoprotein aequorin into the barnacle giant muscle fiber [5]. In the 1980s, Tsien and colleagues produced a variety of fluorescent Ca2+ indicators that provided trustworthy methods for analysis of [Ca2+]i [6]. Since the development of these methods, the study of intracellular Ca2+ signaling was strongly boosted. Overtime, the interests of many researchers shifted from Ca2+ analysis at the cellular level to that of the subcellular level.
It has been found that Ca2+ is not even distributed throughout the whole cell, thus achieving an intracellular heterogeneity of Ca2+. Ca2+ waves and Ca2+ sparks were observed in a variety of living cells, including heart muscle cell and exocrine cells [7,8]. Due to the important role of Ca2+ in biology cell, numerous techniques for analyzing the mechanisms of cellular and/or subcellular Ca2+ activity in living cells have been developed. The most widely used Ca2+ indicators are chemical fluorescent probes because their light signal is quite large for a given change in [Ca2+] compared with other types of Ca2+ indicators. It was developed different fluorescent Ca2+ indicators, and it is important to select the most suitable probe for a given experiment [6]. The former includes indo 1 and fura 2, whereas the latter includes fluo 3, the calcium green class, and rhod 2.
Many molecules change concentration, distribution, and function simultaneously, or sequentially, during numerous intracellular phenomena. Then, the fluorescence microscopy using fluorescent indicators have allowed adequately analyzed of the distribution and dynamics of functional molecules within single intact living cells, including Ca2+ ions. The absorption spectra, including ultraviolet (UV), or visible wavelength (blue, green, and red), of the fluorescent Ca2+ indicator should be evaluated closely, and optimally matched to the maximal output of the excitation light source. In particular, when using single- or multi-photon excitation lasers, the excitation wavelength is fixed so that Ca2+ indicators need to be selected such that the maximum probe absorbance occurs at the available wavelengths of the laser in the system. It has become popular to use multiple dyes to analyze different parameters simultaneously, such as [Ca2+] and pH or [Ca2+] and membrane potential, or to measure [Ca2+] in targeted subcellular components such as mitochondria [9]. Hence, the emission spectra of Ca2+ indicators are the most essential factor in selection of dyes for multiple staining.
The Ca2+-binding affinity, which is reflected by the value of dissociation constant (K d), is one of the most important factor in choosing an adequate Ca2+ indicator [6]. Ca2+ indicators are thought to shift their absorption or emission spectrum, or change their emitted fluorescence intensity in response to Ca2+ at a concentration range between 0.1 ×K d to 10 ×K d [10]. However, Ca2+ sensitivity is generally most reliable in a [Ca2+] range below and very near the K d value [10]. Although high affinity (low K d), Ca2+ indicators may emit bright fluorescence, they may also buffer intracellular Ca2+ [10]. Moreover, because a high-affinity Ca2+ indicator becomes saturated at relatively low [Ca2+], errors in [Ca2+] estimation can occur. Low-affinity (high K d) fluorescent Ca2+ indicators have been developed. One of the uses of these Ca2+ indicators is to measure Ca2+ concentration in subcellular organelles [10]. Because of their low affinity for Ca2+, the kinetic of the reactions is rapid enough to analyze Ca2+ variations accompanying muscle contraction with high temporal resolution [11].
The first-generation fluorescent Ca2+ indicators are represented by Quin 2. Latter, Fura 2 (UV-excited Ca2+ indicator) was developed to allow ratiometric Ca2+ measurement [12]. The maximal absorption peaks are 335 and 363 nm at maximal and minimal [Ca2+]i, respectively, and the emission peak of both, Ca2+-bound and Ca2+-free forms of fura 2, is 500 nm [12]. For ratiometric measurements, excitation at 340 and 380 nm has usually been preferred, because the absorption peak of Ca2+-free fura 2 is quite close to its isobestic point, and Ca2+-free fura 2 emits greater than Ca2+-bound fura 2 when excited by wavelengths longer than 370 nm [12]. Others Ca2+ indicators such as Calcium orange, calcium crimson, fura red, fluo 3 were developed to analysis of intracellular Ca2+ signaling [13].
Rhod 2 is a rhodamine-based Ca2+ indicator produced by Tsien and co-workers [14]. This Ca2+ indicator has been used for measurement of Ca2+ mitochondrial in various cell types, including cardiac myocytes [15]. Because the mitochondria exhibit a large potential difference (inside negative) across its membrane, the acetoxymethyl ester (AM) form of rhod 2, which is a multivalent cation, is effectively accumulated, hydrolyzed, and trapped in mitochondria as is the case with rhodamine-123 or tetramethylrhodamine methyl ester (TMRM) [15]. Due to the excitation/emission spectra of rhod 2 is shifted to the red compared with Ca2+ indicators based on fluorescein, rhod 2 enables the measurement of both cytosolic and mitochondrial [Ca2+] in a single cell in combination with shorter wavelength Ca2+ indicators [15].
It is important to note that the most chemical fluorescent indicators are cell impermeant. Then, to load the living cells with these fluorescent Ca2+ indicators, it is necessary to adopt special invasive or biochemical techniques. Many of the fluorescent Ca2+ indicators are derivatized with an AM that is cell permeable [16]. Unlike the original active form, the AM form of the fluorescent Ca2+ indicators can passively diffuse across cell membranes, and once inside the cell, esterases cleave the AM group of the probe leading to a cell-impermeant indicator [16]. The final intracellular concentration of the hydrolyzed Ca2+indicators is dependent on numerous factors, including the type of Ca2+ indicators and loaded cells, loading concentrations of Ca2+indicators, number of the loaded cells, loading time, loading temperature, and preloading condition [16].
For in vitro quantitative evaluation of [Ca2+] from fluorescence intensity measurements, calibration must be undertaken. It is important to note that the K d value of Ca2+ for the fluorescent probe can be drastically affected by probe-environmental conditions [12]. Thus, it is important to estimate the K d under the experimental conditions [12]. There are two types of the calibration: (1) in vitro calibration and (2) in vivo (in situ) calibration [12]. The intracellular Ca2+ concentration is related to the measured fluorescence intensity by [Ca2+] = Kd × (F−Fmin)/(Fmax−F) [Ca2+] = Kd × (F−Fmin)/(Fmax−F) [Equation 1 - single wavelength measurement] or [Ca2+] = β × Kd × (R−Rmin)/(Rmax−R) [Ca2+] = β × Kd × (R−Rmin)/(Rmax−R) [Equation 2 – ratiometric measurement), where F and R are the experimentally measured fluorescence intensities, and the ratio of these intensities, respectively, Fmin and Rmin are the measured fluorescence intensity, and ratio in the absence of Ca2+, respectively; Fmax and Rmax are measured fluorescence intensity, and ratio of Ca2+-saturated dye, respectively; β is the ratio of the fluorescence intensities at the wavelength chosen for the denominator of R (e.g., 380 nm excitation for fura 2) in zero and saturating [Ca2+]; and K d value is the dissociation constant of the indicator for Ca2+ [12].
The Kd value of most fluorescent Ca2+ indicators estimated using in vitro calibration is not the same as the actual K d in the cell. This discrepancy is dependent on a fact that the K d of the cell contained fluorescent Ca2+ indicator is affected by the temperature, pH, viscosity, ionic strength, and other intracellular constituents [12,16]. Then, the K d measured inside one cell type may not be valid for other cells. In addition, intracellular environments also affect spectral properties of some fluorescent Ca2+ indicators [12,16]. As previously discussed in Equations 1 and 2, the [Ca2+] can be estimated from measurements of the Ca2+ indicator fluorescence at minimal and maximal [Ca2+] [12]. In the case of ratiometric measurements, Rmax and Rmin can be obtained by using low concentrations of fluorescent Ca2+ indicators because the ratio is theoretically independent of the dye concentration [12]. In the case of single-wavelength measurements, however, Fmax and Fmin cannot be obtained unless the dye concentration is precisely estimated, or in vivo calibration is performed in the same cell [12].
The modern fluorescence microscopy techniques combine the power of high performance optical components with computerized control of the instrument, and digital image acquisition, to achieve a level of sophistication that far exceeds that of simple observation by the human eye. Microscopy now depends heavily on electronic imaging to rapidly acquire information at low light levels, or at visually undetectable wavelengths. These technical improvements are not mere window dressing, but are essential components of the light microscope as a system. The microscope accomplishes this first step in conjunction with electronic detectors, image processors, and display devices that can be viewed as extensions of the imaging system. Computerized control of focus, stage position, optical components, shutters, filters, and detectors is in widespread use, and enables experimental manipulations that were not humanly possible with mechanical microscopes. The increasing application of electro-optics in fluorescence microscopy has led to the development of optical tweezers capable of manipulating subcellular structures or particles, the imaging of single molecules, and a wide range of sophisticated spectroscopic applications. The fluorescence microscopy techniques using Ca2+ indicators have been developed to precisely analyze intracellular Ca2+ signaling in living cells. We have used this technique to study the precise role of the intracellular Ca2+ signaling in different physiological and pathological process, including in cardiovascular diseases. In this review, we focused the importance of this technique to biomedical studies on the role of intracellular Ca2+ signaling in the systemic arterial hypertension.
Importance of fluorescence microscopy in the study of the dysfunctions of intracellular Ca2+ homeostasis involved in pathogenesis of cardiovascular diseases
As previously mentioned, Ca2+ is a basic requirement cellular function. This ion participates on the regulation of several physiological cellular functions in all living cells. However, the dysregulation of its intracellular homeostasis can trigger pathological process, including the cell injury and death. We have used this technique to study the precise role of the intracellular Ca2+ signaling in the systemic arterial hypertension. In a study published in 2008 in the Journal of Pharmacology and Experimental Therapeutics, we showed by high resolution amperometry techniques that catecholamine secretion induced by nicotine cholinergic agonist acetylcholine (ACh, 1 mM, 2-s pulse) and depolarizing agent potassium (K+, 70 mM, 2-s pulse) in isolated adrenal chromaffin cells from animal model of systemic arterial hypertension (Spontaneously Hypertensive Rats or SHR) was significantly higher compared with its respective normotensive control rats (NWR) [17]. In relation to NWR, the secretory responses of the adrenal chromaffin cells from SHR have the following characteristics: 1) double number of secretory events, 2) 4-fold augmentation of total secretion, 3) cumulative secretion that saturated slowly, 4) 3-fold higher complex events with two to four superimposed spikes that may be explained by faster spike kinetics, 5) about 2- to 3-fold higher event frequency at earlier post stimulation periods, and 6) 2- to 5-fold higher quantal content of simple spikes. We conclude that SHR cells have faster and larger catecholamine release responses, explained by more vesicles ready to undergo exocytosis and greater quantal content of vesicles [17]. As Ca2+ is a basic requirement for neurotransmitter/hormone exocytosis, we postulated that a dysregulation of intracellular Ca2+ homeostasis in adrenal chromaffin cells could be involved in the catecholamine hypersecretion in hypertensives. Due to involvement of sympathetic dysfunctions in the systemic arterial hypertension, these findings could have relevance to further understand the pathogenic mechanisms involved in the development of hypertension, as well as in the identification of new drug targets useful to the antihypertensive therapy.
To test the hypothesis of involvement of the dysregulation of intracellular Ca2+ homeostasis in adrenal chromaffin cells in the catecholamine hypersecretion in hypertension, we used the fluorescence microscopy techniques previously mentioned to study the intracellular Ca2+ signaling in the slices of adrenal medulla from hypertensive and normotensive animals [18,19].
To study the changes of the cytosolic Ca2+ concentration ([Ca2+]c), slices were incubated with 10 μM of the ratiometric dye fura-2AM in a Tyrode solution containing 0.01% pluronic acid F-127 at room temperature for 45 min [12]. This solution was oxygenated and protected from light. After incubation, slices were washed and mounted in a Leiden chamber with 1 ml of Tyrode solution and kept at 37°C during the experiments. Basal [Ca2+]c was measured during the first 10 images, which were taken at 5-s intervals. Calibration was done by adding 1 mM digitonin (maximal fura-2 fluorescence, Rmax). [Ca2+]c elevations in the chromaffin cells from adrenal medulla slices in response to activation of the nicotine cholinoceptors by acetylcholine (ACh) and membrane depolarization by potassium (K+) were studied. All experiments began with an initial 5-min perifusion resting period to adapt the slice to its new environment. Once the basal fluorescence was stable, the normal Tyrode solution containing 1.8 mM Ca2+ was switched to another containing ACh (100 μM) or K+ (70 mM) that bathed the slice during 80 s.
Figure 1A shows fluorescence example images from a NWR in basal conditions, after challenging with ACh, and the final maximal fluorescence elicited by digitonin (panels 1, 2, and 3, respectively). The bottom panels show the fluorescence signal produced in a SHR slice, that was more intense under the three experimental conditions i.e., basal, ACh, and digitonin (panels 4, 5 and 6), in comparison with the NWR slice. Figure 1B shows representative records of the time courses of [Ca2+]c elevations (measured as normalized fluorescence ratios at 340/380 wavelenghts) elicited by ACh in a NWR slice and a SHR slice. The responses had two phases namely, an initial transient peak that was followed by a decay phase to reach a plateau. The amplitude of the peak, and the plateau, were measured as indicated by double head arrows. Although the two responses were qualitatively similar, they however exhibited clear quantitative differences, as shown in panels C and D of figure 1. For instance, the peak [Ca2+]c was 60% higher in SHR and the plateau 101% greater, as compared with NWR. The time constants for activation (τa) and inactivation to a plateau (τi) of the [Ca2+]c transients also were evaluated. So, τa were 25 s and 20 s and τi were 25 s and 35 s, respectively in NWR and SHR. Hence, τa was 20% lower in SHR and τi was 39% higher in SHR, as compared with NWR (Figure 1D).
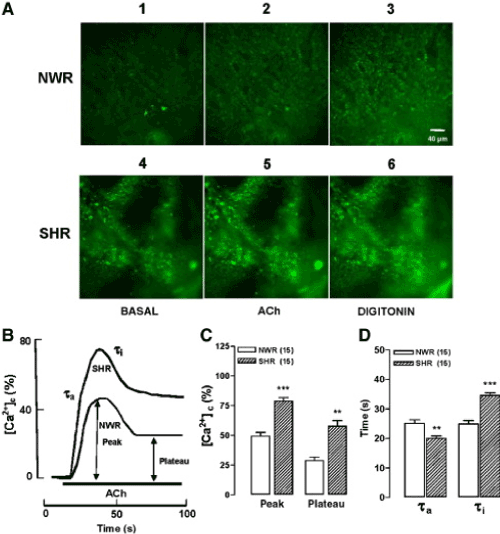
Figure 1. ACh-stimulated [Ca2+]c elevations were faster and higher in adrenal slices of spontaneously hypertensive rats (SHR), as compared with its respective normotensive control rats (NWR). Slices were loaded with Fura-2AM and were continuously perifused with an oxygenated (95% O2 and 5% CO2) Tyrode solution containing 1.8 mM Ca2+. Slices were stimulated with cholinergic agonist ACh (100 μM) for 80 s. A. Digital images showing the Fura-2 fluorescence under basal, ACh, or digitonin treatment conditions (panels 1, 2 and 3 for NWR and 4, 5 and 6 for SHR slices). B. Typical records representing the fluorescence of Fura-2 (R340nm/380nm, normalized as % of maximal fluorescence) in NWR and SHR slices stimulated with ACh (bottom horizontal bar). C. Averaged results from 15 NWR and SHR slices. D. Time constants for activation (τa) and inactivation to a plateau (τi) of the [Ca2+]c transients. The parameters plotted in C and D were calculated from experiments of the type shown in panel B. Data are means ± SEM, showing differences for SHR (*P < 0.05 and ***P < 0.001) compared with NWR. Extracted from Miranda-Ferreira et al. [19].
A picture similarly to that for ACh was found in slices stimulated with a Tyrode solution containing 70 mM K+ (low Na+). In the representative fluorescence micrographs shown in figure 2A, fluorescence intensities were substantially higher in SHR slices (compare panels 1, 2 and 3 with panels 4, 5 and 6) under the three experimental conditions (basal, K+, and digitonin). The time courses of the changes in fluorescence elicited by K+ are shown in figure 2B; the [Ca2+]c peak was higher and sharper in SHR, compared with NWR. Again, the [Ca2+]c declined to a plateau in both preparations. The bar graph of figure 2C represents averaged results obtained in 15 slices from NWR and SHR challenged with K+. The fluorescence peak was 44% higher and the plateau 88% higher in SHR, compared with NWR. As in the case of ACh, the [Ca2+]c time course curves showed differences in their time constant for activation and inactivation. Thus, τa was 20 s and 14 s, and the τi was 28 s and 45 s, respectively in NWRs and SHRs. So, the τa was 27% lower in SHR and τi was 59% higher in SHR, as compared with NWR slices (Figure 2D).
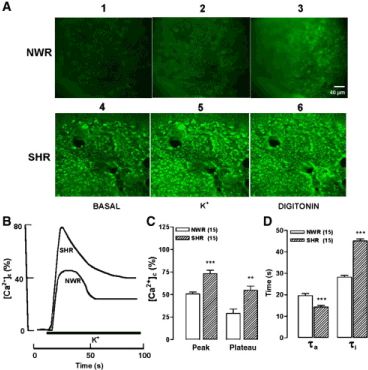
Figure 2. Stimulation with a high-K+ solution produced [Ca2+]c elevations that were faster and higher in adrenal slices of SHR, as compared with NWR. After a 5-min pre-equilibration period, slices were challenged with 70 mM K+ during 80 s. A. Digital images showing the Fura-2 fluorescence under basal, K+, or digitonin treatment conditions for NWR (panels 1, 2 and 3) and SHRs (panels 4, 5 and 6). B. Typical records representing the time course of Fura-2 fluorescence of (R340/380, normalized as % of maximal fluorescence) in NWR and SHR slices stimulated with K+ during the time period shown by the bottom horizontal bar. C. Averaged results from 15 NWR and 15 SHR slices. D. Time constants for activation (τa) and inactivation to a plateau (τi) of the [Ca2+]c transients. The parameters plotted in C and D were calculated in each individual slice, from traces similar to those shown in panel B. Data are means ± SEM for SHR, showing differences (**P < 0.01, ***P < 0.001) with respect to NWR. Extracted from Miranda-Ferreira et al. [19].
To study Ca2+ movements in the mitochondrial compartment, slices were incubated with Rhod-2AM (10 μM), an indicator that presents a higher affinity for mitochondria. Slices were incubated in a Tyrode solution containing Rhod-2.AM plus 0.01% pluronic acid F-127 at 37°C for 60 min, oxygenated and protected from light. After incubation with Rhod-2, slices were bathed with Ca2+-free medium and digitonin (5 µM, 30 s) to produce a semi-permeabilization of the plasma membrane to assure the washout of the indicator not confined to the mitochondrial compartment. The Ca2+-free medium (intracellular medium) contained (in mM): 35 NaCl, 115 KCl, 1 KH2PO4, 20 HEPES, 2 EGTA, pH 7.2; this solution was supplemented with 1 mM ATP, 10 μM creatine phosphate, 10 μM creatine phosphate kinase, and 2 mM pyruvate to restore the energy expenditure caused by the cell semi-permeabilisation. Calibration was done by addition of 1 mM ionomycin, a membrane permeant that allows finding out minimal fluorescence at the end of the experiment (Rmin). To study the changes of the mitochondrial Ca2+ concentration ([Ca2+]m), the effects of the protonophore FCCP (carbonylcyanide p-(trifluoromethoxy) phenylhydrazone, 5 μM) were evaluated. FCCP stimulates the release of Ca2+ from mitochondria. We also studied the cross-talking of mitochondria and the endoplasmic reticulum by using a mixture containing caffeine plus ryanodine plus thapsigargin, named CRT, to stimulate the leak of Ca2+ from endoplasmic reticulum and observe its uptake into mitochondria. Conversely, we also studied how the mitochondrial Ca2+ released by FCCP was taken up by the endoplasmic reticulum.
The FCCP dissipates the H+ gradient and causes mitochondrial depolarization. This leads to interruption of Ca2+ uptake and the release into the cytosol of mitochondrial Ca2+, thus explaining the [Ca2+]c elevation produced by FCCP in the experiments shown in Figure 3C and 3D. A more direct measure of [Ca2+]m changes elicited by FCCP was attempted using the mitochondrial Ca2+ indicator Rhod-2. Figure 3A shows fluorescence images taken from a NWR slice (panels 1–4) or a SHR slice (panels 5–8). The basal fluorescence was considerably higher in SHR, as compared with NWR (panels 1 and 5). This difference was more than double, as indicated in the bar graph of figure 1. Also, fluorescence was more intense in SHR slices treated with 5 and 10 μM FCCP, compared with NWR slices.
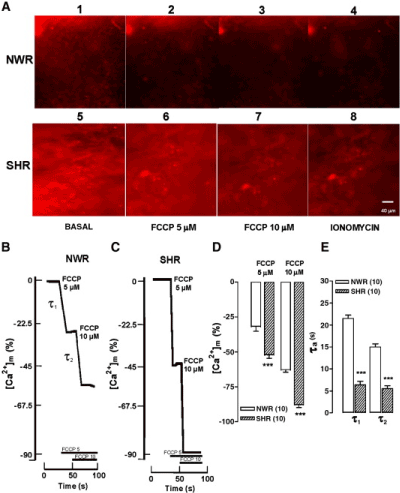
Figure 3. FCCP causes a greater and faster Ca2+ release from mitochondria of SHR adrenal slices, as compared with NWR slices. Slices loaded with the mitochondrial Ca2+ fluorescence indicator Rhod-2 were continuously perifused with an oxygenated Tyrode solution containing 1.8 mM Ca2+. After a 5-min equilibration period, they were exposed to the protonophore FCCP at the indicated concentrations. A. Panels 1–4 and 5–8 show the fluorescence images taken just before (basal) or at the end of FCCP (5 or 10 μM) or ionomycin (1 μM) additions. B and C, traces showing the time course of the [Ca2+]m decay, obtained from slices of NWR (panel B) and SHR adrenals (panel C). D. Pooled data on the extent of [Ca2+]m depletion elicited by the two concentrations of FCCP. E. Time constants for the rates of [Ca2+]m decay upon FCCP treatment. Data in panels D and E are means ± S.E.M. of 10 experiments. ***P < 0.001, SHR compared with NWR. Extracted from Miranda-Ferreira et al. [19].
Figure 3B displays an original trace of the [Ca2+]m changes elicited by two concentrations of FCCP added sequentially to the perifusion solution. Upon addition of 5 μM FCCP, [Ca2+]m decayed quickly to reach a new steady state level at 30% of the initial. Then, the subsequent addition of 10 μM FCCP produced further Ca2+ loss that decayed until 52% of the initial value. The effects of FCCP were faster and higher in the representative records on [Ca2+]m obtained from a SHR slice (Figure 3C). At 5 μM, FCCP caused a 52% Ca2+ depletion and at 10 μM the [Ca2+]m decayed by almost 90%. The decay was higher in SHR, as compared with NWR slices. Figure 3D summarizes the effects of FCCP on [Ca2+]m, which caused a 62% (FCCP 5 μM) and 40% (FCCP 10 μM) greater Ca2+ release in SHR, as compared with NWR slices. The Ca2+ decay time constants were 69% smaller at 5 μM FCCP and 60% smaller at 10 μM FCCP in SHR, compared with NWR slices. In summary, FCCP caused a greater and faster Ca2+ release from mitochondria of SHR chromaffin cells, compared with NWR.
In summary, these findings suggest that adrenal chromaffin cells from hypertensives generate a substantially greater peak and plateau elevations of [Ca2+]c and [Ca2+]m, as compared with the responses of chromaffin cells from normotensives. Furthermore, activation of Ca2+ transients was faster and inactivation slower in hypertensives. As Ca2+ is a basic requirement for neurotransmitter/hormone exocytosis, these changes of Ca2+ homeostatic mechanisms could explain the greater quantal catecholamine release responses observed in hypertensives, as compared with normotensives adrenal chromaffin cells [17,18]. Then, this dysregulation of intracellular Ca2+ homeostasis in adrenal chromaffin cells could be involved in the pathogenesis of systemic arterial hypertension. Due to involvement of these sympathetic dysfunctions in the hypertension, these findings could have relevance to further understand the pathogenic mechanisms implicated in the development of this cardiovascular disease, as well as in the identification of new drug targets useful to the antihypertensive therapy.
Conclusion
The fluorescence microscopy techniques using fluorescent Ca2+ indicators have been developed to precisely analyze intracellular Ca2+ signaling in living cells. The use of these techniques to study the precise role of the intracellular Ca2+ signaling in different physiological and pathological process, such as cardiovascular diseases, can contribute decisively for understanding of the role of intracellular Ca2+ signaling in the pathophysiology of hypertension, and development of more efficient and safer pharmacological antihypertensive strategies to treat human hypertension.
Acknowledgment
Our studies were supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Disclosure
The authors report no conflicts of interest.
References
- Berridge MJ (1993) Inositol trisphosphate and calcium signalling. Nature. 361: 315-325. [Crossref]
- Ringer S (1882) Concerning the Influence exerted by each of the Constituents of the Blood on the Contraction of the Ventricle. J Physiol 3: 380-393. [Crossref]
- Heilbrunn LV, Wiercinski FJ (1947) The action of various cations on muscle protoplasm. J Cell Comp Physiol 29: 15-32. [Crossref]
- EBASHI S (1961) Calcium binding activity of vesicular relaxing factor. J Chir (Paris) 82: 236-244. [Crossref]
- Ridgway EB, Ashley CC (1967) Calcium transients in single muscle fibers. Biochem Biophys Res Commun 29: 229-234. [Crossref]
- Williams DA, Fogarty KE, Tsien RY, Fay FS (1985) Calcium gradients in single smooth muscle cells revealed by the digital imaging microscope using Fura-2. Nature. 318: 558-561.
- Takamatsu T, Wier WG (1990) Calcium waves in mammalian heart: quantification of origin, magnitude, waveform, and velocity. FASEB J. 4: 1519–1525.
- Petersen OH, Petersen CC, Kasai H (1994) Calcium and hormone action. Annu Rev Physiol 56: 297-319. [Crossref]
- Zorec R, Hoyland J, Mason WT (1993) Simultaneous measurements of cytosolic pH and calcium interactions in bovine lactotrophs using optical probes and four-wavelength quantitative video microscopy. Pflügers Arch 423: 41-50.
- Meyer T, Wensel T, Stryer L (1990) Kinetics of calcium channel opening by inositol 1,4,5-trisphosphate. Biochemistry 29: 32-37. [Crossref]
- Escobar AL, Monck JR, Fernandez JM, Vergara JL (1994) Localization of the site of Ca2+ release at the level of single sarcomere in skeletal muscle fibers. Nature 367: 739-741.
- Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440-3450.
- Lipp P, Niggli E (1993) Ratiometric confocal Ca2+-measurements with visible wavelength indicators in isolated cardiac myocytes. Cell Calcium 14: 359-372.
- Minta A, Kao JP, Tsien RY (1989) Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J Biol Chem 264: 8171-8178. [Crossref]
- Trollinger DR, Cascio WE, Lemasters JJ (1997) Selective loading of rhod-2 into mitochondria shows mitochondrial Ca2+ transients during the contractile cycle in adult rabbit cardiac myocytes. Biochem Biophys Res Commun 236: 738-742.
- Tsien RY (1981) A non-disruptive technique for loading calcium buffers and indicators into cells. Nature 290: 527-528. [Crossref]
- Miranda-Ferreira R, de Pascual R, de Diego AM, Caricati-Neto A, Gandía L, et al. (2008) Single-vesicle catecholamine release has greater quantal content and faster kinetics in chromaffin cells from hypertensive, as compared with normotensive, rats. J Pharmacol Exp Ther 324: 685-693.
- Miranda-Ferreira R, de Pascual R, Caricati-Neto A, Gandía L, Jurkiewicz A, et al. (2009) Role of the endoplasmic reticulum and mitochondria on quantal catecholamine release from chromaffin cells of control and hypertensive rats. J Pharmacol Exp Ther 329: 231-40.
- Miranda-Ferreira R, de Pascual R, Smaili SS, Caricati-Neto A, Gandía L, et al. (2010) Greater cytosolic and mitochondrial calcium transients in adrenal medullary slices of hypertensive, compared with normotensive rats. Eur J Pharmacol. 636: 126-136.