Bullous congenital ichthyosiform erythroderma (BCIE), also known as epidermolytic hyperkeratosis is a rare autosomal dominant disorder of keratinization, due to gene mutation in keratine 1 and 10, with a prevalence of 1 in 200,000 to 300,000 people. However, half of cases occur sporadically as the result of a new mutation. The diagnosis is clinical but needs to be confirmed with histopathology in some doubtful cases. It is characterized by blistering and erythroderma in early life with many similarities to epidermolysis bullosa simplex, but with subsequent development of ichthyosis and hyperkeratosis throughout life. The histologic features of this disease are characteristic but not specific. Clinical management consists of a symptomatic treatment because a cure is not yet possible. We report a rare case of a 6-year old child with sporadic BCIE present since birth. Through our case, we highlight the clinical and histological aspect of this rare pathology, with special concern of the therapeutic options especially in children
Bullous congenital ichthyosiform erythroderma, epidermolytic hyperkeratosis, genodermatosis, ichtyosis, bullous disorder, retinoids
Bullous congenital ichthyosiform erythroderma (BCIE), also known as epidermolytic hyperkeratosis is a rare autosomal dominant disorder of keratinization [1] that often presents at birth with generalized erythema, scales, blistering, and erosions [2]. However, half of cases occur sporadically as the result of a new mutation [3]. This genodermatosis is generally caused by mutation in epidermal keratins (KRT) 1 and 10 [4]. It is characterized by blistering and erythroderma in early life with many similarities to epidermolysis bullosa simplex [5]. However, these patients subsequently develop ichthyosis and hyperkeratosis throughout life [5]. Additionally, it has characteristic histopathologic findings [6]. We report a rare case of a 6-year old child with sporadic BCIE present since birth.
A 6-year old boy, second of a fratery of 4, born of a 1st degree consanguineous parentage and normal full-term delivery, presented with a history of scalded appearance with diffuse shedding of scales at birth. He reported, since the age of 2 months, the appearance of flaccid blisters with clear content on the extension faces of the limbs, extending thereafter to the abdomen and the face, with subsequent development of diffuse erosions. Six months after, the patient developed very itchy, thick and diffuse blackish patches all over the body, increasing in winter, without improvement under topical treatment alone. Otherwise, the patient did not experience a disorder of the growth curve nor a delay of the acquisitions. Additionally, there were no familiar similar cases.
Clinical examination found generalized scaling with multiple erosions having a clean surface and a peripheral collar, with the presence of hemorrhagic crusts in their surface by places, on the extremities of the limbs, the popliteal folds and the face, associated with a diffuse pachydermia all over the body, leading to a limitation of mouth opening. This pachydermia was made of small, non-adherent, dark brown thick scales that were arranged in sometimes in plaques on the folds. Nevertheless, the palms, soles, nails and mucosa were normal. Systemic examination didn’t show any abnormality.
A cutaneous biopsy was made showing a marked compact orthokeratosic hyperkeratosis, acanthosis with mild acantholysis, and hypergranulosis with large dotted cells having a clear cytoplasm and basophilic and eosinophilic granulations in the granular and spinous layer. This indicated the presence of a disorder of maturation and keratinization in favor of congenital ichthyosiform erythroderma.
A pre-retinoid assessment was performed and the patient was treated with acitretin at a dose of 0.3 mg / kg / day in combination with an emollient cream with urea and supergrass gels, with slight clinical improvement.
BCIE, also known as epidermolytic hyperkeratosis (EHK) [4], is a rare genodermatosis with a prevalence of 1 in 200,000 to 300,000 people [7]. It was first described by Brocq in 1902 as an entity distinct from congenital ichthyotic erythroderma, a non-blistering condition [8]. It is an autosomal dominant condition with one hundred percent penetrance, although approximately 50% of all cases represent new spontaneous mutations [6]. KRT 1 and KRT 10 gene mutations represent the underlying cause [2], responsible for the epidermal fragility seen in BCIE [8], with heterozygous point mutations the most frequent (86.6%) although other have also been reported [1].
The diagnosis is clinical but needs to be confirmed with appropriate laboratory investigations [1]. Indeed, histopathology findings help in making a diagnosis especially if there is no history of an affected parent [4]. It usually presents as a bullous disease of the newborn followed by an ichthyotic skin disorder throughout life [6]. However, it is far less common than staphylococcal scalded skin syndrome (SSSS), with which it may be confused at presentation, especially in the absence of a family history [8]. This rare disease is characterized by erythematous, erosive and bullous skin lesions at birth or shortly thereafter [2], with appearance of a widespread hyperkeratosis, that replaces the erythema and bullae, in infancy and into adulthood, leading to a characteristic cobblestone appearance [6]. This hyperkeratosis is made with large, ashy-browne colored scales present at joint flexures, resulting in verrucous or hystrix like macules [2]. Additionally, palms and soles may be involved, thus predecting a KRT 1 gene mutation [4].
This condition should be mainly differentiated, in childhood, from non-bullous congenital ichthyosiform erythroderma (CIE) by the absence of history of bullae and the presence of erythroderma, and from epidermolysis bullosa where the bulla formation predominate at trauma prone areas with the absence of scaling or hyperkeratosis [4]. Other differential diagnosis includes SSSS, lamellar ichtyosis, Omenn syndrome, and toxic epidermal necrolysis [2]. These disorders can usually be distinguished on clinical signs; however, histological examination might be helpful to make the definitive diagnosis [1].
Histopathological findings indicate marked hyperkeratosis, acanthosis and granular degeneration of upper spinous to granular layers of the epidermis creating multiple vacuoles with large keratohyalin granules and intercellular edema [1,2]. These histologic features are characteristic for patients with generalized EHK, Ichthyosis Bullosa of Siemens (IBS) and mosaic EHK, however, they could also be seen in other conditions such as epidermolytic acanthoma and nevoid follicular epidermolytic hyperkeratosis [6], thus making them unspecific for this disorder [1]. Moreover, the boundaries appear to be formed by eosinophilic and basophilic granules, by light microscopy [6], with the appearance of complex aggregates of KRTs in the upper spinous to granular layers under electron microscopy [2]. Additionally, immunohistochemical staining can be used to show a defect in expression of KRT 1 or KRT 10 [1].
BCIE is a strikingly heterogenous disorder with widely varying severity [1]. In fact as the patient’s age, the scaling becomes thicker and the propensity to blister decreases [4]. Furthermore, erythroderma is lessening with age [2].
Clinical management consists of symptom reduction because a cure is not yet possible [3]. Treatment in early period is directed towards treating secondary complications of erosions, mainly sepsis and electrolyte imbalance [4]. In newborns, an intensive care nursery in highly humidified incubators should be considered, and if necessary, systemic antibiotic treatment can be initiated [1]. In the other hand, the management of older patients with hyperkeratosis requires regular emollients, keratolytics, antiseptic washes and systemic antibiotics as necessary [8]. In severely affected patients, systemic and topical retinoids are effective in reducing hyperkeratosis [3]. In fact, systemic retinoids such as acitretin (usually at doses of 0.2–0.5 mg/kg daily) or etretinate promote desquamation, which helps some children and adults but causes excessive fragility in others [8]. However, patients with mutations in KRT10 seems to respond better to retinoid therapy than patient with KRT1 mutation [1]. Thus, gene therapy for instance with topical application of siRNA or similar agents may become feasible in the future (Figures 1 and 2) [1].
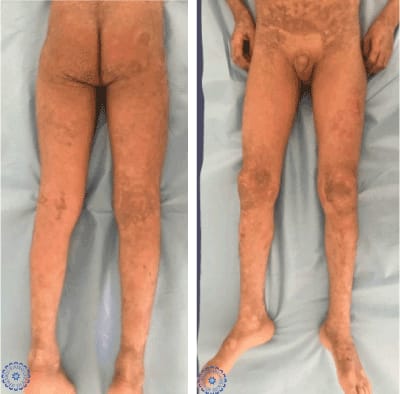
Figure 1. Clinical images showing a diffuse pachydermia of the trunk with some erosions on the back covered by hemorrhagic crusts.
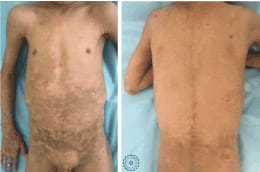
Figure 2. Clinical images showing diffuse pachydermia of the lower extremities with large brown scals and fissures.
To sum up, we report a rare sporadic case of BCIE in a 6-year old child. Through our case, we highlight the importance of careful clinical examination for the diagnostic orientation of BCIE, in addition to the role of histopathology to help making the diagnosis and rule out other conditions. We also emphasize the importance of regular follow-up and age-appropriate management of the patient, while paying particular attention to the use of systemic retinoids given the risk of complications.
- Kucharekova M, Mosterd K, Winnepenninckx V, van Geel M, Sommer A, et al. (2007) Bullous congenital ichthyosiform erythroderma of Brocq. Int J Dermatol 46: 36-8. [Crossref]
- Kurosawa M, Takagi A, Tamakoshi A, Kawamura T, Inaba Y, et al. (2013) Epidemiology and clinical characteristics of bullous congenital ichthyosiform erythroderma (keratinolytic ichthyosis) in Japan: Results from a nationwide survey. J Am Acad Dermatol 68: 278-83. [Crossref]
- Betlloch I, Lucas Costa A, Mataix J, Pérez-Crespo M, Ballester I (2009) Bullous congenital ichtyosiform erythroderma: a sporadic case produced by a new KRT10 gene mutation. Pediatr Dermatol 26: 489-91. [Crossref]
- Mondal AK, Kumar P, Mondal A (2011) Bullous congenital ichthyosiform erythroderma. Indian Pediatr 48: 968.
- Jha A, Taneja J, Ramesh V, Singh A (2015) Annular epidermolytic ichthyosis: A rare phenotypic variant of bullous congenital ichthyosiform erythroderma. Indian J Dermatol Venereol Leprol 81: 194-7. [Crossref]
- Ross R, DiGiovanna JJ, Capaldi L, Argenyi Z, Fleckman P, et al. (2008) Histopathologic Characterization of Epidermolytic Hyperkeratosis: A Systematic Review of Histology from the National Registry for Ichthyosis and Related Skin Disorders. J Am Acad Dermatol 59: 86-90. [Crossref]
- El-Khateeb EA (2008) Bullous congenital ichtyosiform erythroderma associated with hypocalcemic vitamin D-resistant rickets. Pediatr Dermatol 25: 279-82.
- Cheng S, Moss C, Upton CJ, Levell NJ (2009) Bullous congenital ichthyosiform erythroderma clinically resembling neonatal staphylococcal scalded skin syndrome. Clin Exp Dermatol 34: 747-8. [Crossref]