Abstract
The modulation of iron homeostasis is essential for the survival of humans. Iron supports many vital biological processes including oxygen transport and storage, DNA synthesis, metabolic energy, and cellular respiration Derangement in iron homeostasis leads to iron overload or iron deficiency, which are involved in a variety of pathophysiologic conditions including anemia and iron-overload related disorders. Iron overload causes the accumulation of iron mainly in the heart, liver and endocrine glands causing damages to these organs. The accumulation of excess iron on the myocardium may cause iron-overload (or siderotic) cardiomyopathy (IOC), a leading cause of death in transfusion-dependent patients. Currently, IOC has limited therapies because of the lack of valid clinical models for detection of sub-clinical IOC. The present review seeks to consolidate current research findings on IOC to advance clinical knowledge and understanding of its definition, etiology, clinical manifestation, pathogenesis, diagnosis and clinical management. Included in the present review are two meta-analyses of diagnostic methods and clinical management approaches of IOC.
Key words
cardiomyopathy, diagnosis, Cardiac amyloidosis
Introduction
The earliest mention of the association between altered iron homeostasis and heart failure in medical literature was in mid-19th Century linked with iron deficiency [1-6]. It was observed that an estimated 25% of patients receiving repeated therapeutic phlebotomy – a prophylactic or curative therapy involving withdrawal of blood from a patient to treat various medical conditions associated with the accumulation of excess iron in the body – developed iron deficiency anemia and overt heart failure [7]. However, the recognition of the deleterious effect of iron overload on cardiac function was much more recent, first proposed in 1981 by Sullivan in a series of studies investigating the relationship between iron and ischemic heart diseases [8-11]. Heart diseases had a significant gender bias, more prevalent in males. In addition, menstruating women had very low rates of myocardial infarction (MI), which was associated with low levels of stored iron compared to men and women before the age of 20 years. Monthly menstruation was believed to rid the body of approximately 180-360 mg of iron per year. Further, when menstruation ceases, the rate of heart disease increases promptly [9,10]. The relationship between rising levels of stored iron and heart diseases at a younger age in males and in later years in females suggested iron overload could have a central role in the development of heart disease [12]. Several current studies have established iron overload as a cause of myocardial dysfunction, clinically referred to as iron-overload cardiomyopathy (IOC) [13-16].
Cardiomyopathy define a heterogeneous group of myocardial disorders classified as either primary or secondary. Primary cardiomyopathies are often of idiopathic (unknown) causes while secondary cardiomyopathies occur in the setting of demonstrable cardiac or extra cardiac causes [12]. Iron overload cardiomyopathy (IOC) is a recently identified secondary form of cardiomyopathy occurring in the setting of accumulation of iron on the myocardium. Recently, IOC has attracted increased research interest primarily because iron overload is a frequently encountered condition in certain hematologic conditions and secondarily because of improvements in diagnostic accuracy and therapeutic efficacy [12-14]. Despite increased recognition, the current classification systems of cardiomyopathies based on morphological and functional myocardial features fail to recognize IOC as a distinct form of cardiomyopathy. The 2016 position statement of the European Society of Cardiology (ESC) Working Group on Myocardial and Pericardial Diseases classifies IOC as one of etiologies of dilated cardiomyopathy (DCM) resulting from chronic blood transfusion and haemochromatosis [17]. The 2006 American Heart Association (AHA) Scientific Statement on Contemporary Definitions and Classification of the Cardiomyopathies also recognize IOC as a sub-type of DCM occurring in the setting of abnormal iron absorption or high volume blood transfusion [18-19].
The current definitions of IOC focus on either etiologic or clinicopathologic factors (clinical signs and symptoms). The AHA provides an etiology-based definition, describing IOC as iron overload of the cardiac myocytes occurring in the setting of abnormal iron absorption such as hereditary hemochromatosis (HH) or because of high volume blood or parenteral iron infusions [19]. Other definitions focus on clinicopathologic factors, describing IOC as a form of dilated cardiomyopathy characterized by left ventricular (LV) remodeling with chamber dilation and reduced LV ejection fraction (LVEF) [5]. IOC has also been defined as the presence of systolic or diastolic cardiac dysfunction secondary to increased accumulation of iron in the myocardium independent of other concomitant processes [20,21]. Drawn from the current definitions, IOC is a secondary form of dilated cardiomyopathy (DCM) resulting from iron deposition in the cardiomyocytes due to genetically determined disorders of iron metabolism or chronic blood transfusion leading to both or either diastolic and/or systolic dysfunction.
The principal etiology of IOC is the deposition of excess iron in the cardiomyocytes. The main causes of excess iron in the body is genetic disorders of iron metabolism such as haemochromatosis (increased gastrointestinal [GI] absorption) and thalassemia or excessive administration of exogenous iron through dietary sources or red blood cells (RBCs) transfusions (hemosiderosis) [5]. Table 1 summarizes the common primary and secondary etiologies of IOC alongside their mechanisms and organs at risk of iron deposition [5].
Table 1: Etiological Conditions of Iron Overload Cardiomyopathy
Iron Deposition |
Condition |
Mechanism |
Primary Etiologies: Hereditary Hemochromatosis |
Type I (mutation in HFE gene): Classical HH |
Increased GI absorption with normal diet. |
Liver, heart endocrine glands |
Type II (Juvenile hemochromatosis) |
Increased GI absorption with normal diet. |
Liver, heart endocrine glands |
Type III |
Increased GI absorption with normal diet. |
Liver, heart endocrine glands |
Type IV |
Increased GI absorption with normal diet. |
Macrophages, liver, heart,
endocrine glands |
a) Iron-Loading Anemia (transfusion-related) |
|
Secondary Etiologies (Conditions) |
Thalassemia |
Increased GI absorption |
|
Sickle Cell Anemia |
Transfusion-related |
Heart, liver, pancreas, pituitary |
Sideroblastic anemia |
Increased GI absorption with normal diet. |
Heart, Liver |
Diamond-Blackfan Anemia |
Transfusion-related |
Heart, neurons, mitochondria |
Post-stem cell transplant |
Transfusion-related |
Heart, liver |
Chronic Kidney Disease/End stage renal failure/dialysis |
Oral and IV iron supplementation |
Heart, liver |
b) Dietary Overload |
Increased dietary intake |
Heart, liver |
|
|
Heart, liver, endocrine |
|
|
The primary etiology of iron overload is hereditary hemochromatosis (HH): an autosomal disorder caused by mutations in the genes encoding proteins involved in iron metabolism. The disorder causes iron overload due to increased gastro-intestinal iron absorption and altered iron metabolism [5]. Four types of HH have been identified based on the type of gene mutation gene. Table 2 provides a summary of the four HH types, inheritance pattern, the main genes and chromosomes involved alongside the type of protein encoded [14].
Table 2: Types of Hereditary Hemochromatosis Causing Iron Overload Cardiomyopathy.
HH Type |
Inheritance |
Gene |
Chromosome |
Encodes |
Type 1 |
Autosomal Recessive |
HFE gene |
Chromosome 6 |
|
Type 2 |
Autosomal Recessive |
HJV gene |
Chromosome 1 |
Hemojuvelin |
Autosomal Recessive |
HAMP gene |
Chromosome 19 |
Hepcidin |
Type 3 |
Autosomal Recessive |
TfR2 gene |
Chromosome 7 |
Transferrin Receptor 2 |
Type 4 |
Autosomal Dominant |
SLC40A1 gene |
Chromosome 2 |
Ferroportin |
In Table 2, the first three types (1 to 3) of HH (mutations) are inherited in an autosomal recessive (AR) pattern while Type 4 HH is inherited in an autosomal dominant (AD) pattern. Types 1, 3, and 4 clinically manifest in adulthood at the fourth or fifth decade of life while Type 2, referred to as juvenile HH manifests much earlier, in the second or third decade of life, and is considered the most severe phenotype [24]. The typical clinical trial of HH is cirrhosis, bronze skin and diabetes mellitus (DM). However, the phenotype of HH varies extremely and depends on several interfering genetic and other factors especially in classical HH (Type 1) [24,25]. In Type 1 and 3, hepatic involvement predominates while in Type 2 endocrine and cardiac complications are more pronounced with HF a frequent cause of death before the third decade of life [24-26]. Cardiac involvement occurs in about 35% of HH cases. Classical HH is prevalent in individuals with Northern European Ancestry and it is one of the most common genetic disorder among whites with an incidence rate of 1 in 500 people in America [19].
Secondary etiologies of IOC refer to accumulation of iron in the cardiomyocytes due to exogenous sources. The primary channels for secondary excessive iron and accumulation are parenteral iron administration and excess administration of exogenous iron.
Oral iron administration is the preferred and safest first-line therapy for many patients with iron deficiency anemia. However, for patients intolerant or unresponsive to oral iron therapy, parenteral iron administration is considered [27]. In patients with hereditary anemias such as thalassemia and sickle cell anemia and acquired anemia such as myelodysplastic syndromes (MDS), Myelofibrosis, aplastic anemia, Sideroblastic anemia, and Blackfan-Diamond anemia, blood transfusion is the cornerstone of treatment [28]. A unit of packed RBCs usually comprises of 200-250 mg of elemental iron, which accumulates in the body because of the lack of an active iron excretion system. Transfusion-dependent patients receive approximately 20 times the normal intake of iron. Thus, high or chronic blood transfusions potentially leads to iron overload with deposition of iron in multiple organs including the heart primarily observed in patients with hereditary anemias [5]. Early detection of hereditary and acquired transfusion-dependent anemias is associated with decreased mortality rates due to improved treatment efficacy. However, treatment requires persistent chronic transfusion, which is the key reason for increased incidence of iron overload in these cohorts of patients [29-31]. Whereas improvements in survival have been noted over the past four decades, iron induced cardiotoxicity remains a leading cause of morbidity and mortality especially in patients with HH and Thalassemia major, and once heart failure develops, prognosis if such patients is ominous [5].
Besides parenteral iron administration, significant amounts of exogenous iron occurs through dietary intake. According to Gordeuk et al. [32], Africans have a high dietary iron intake because of drinking traditional beers fermented in steel drums, referred to as African Iron Overload. Although the mechanism of African Iron Overload, was initially believed to be the etiology of hepatic carcinoma and cardiomyopathy in these patients, other reports indicate environmental factors superimposed on genetic predisposition provides a better explanation for the development of the condition [33,34].
Iron overload cardiomyopathy occurs in the primary or secondary form. The secondary form is more frequent in HH. Two phenotypes of IOC have also been identified: the dilated phenotype and the restrictive phenotype. The dilated phenotype is characterized by LV remodeling resulting into chamber dilation and depressed LV ejection fraction (LVEF). The restrictive phenotype is characterized by diastolic LV dysfunction with restrictive filling, preserved LVEF, pulmonary hypertension and subsequent RV dilation [14]. The two phenotypes are followed by several other clinical manifestations including conduction abnormalities, tachyarrhythmias and perimyocarditis [5,35].
The etiologies of IOC have a wide spectrum and variable symptoms. At the onset of the disease, patients are asymptomatic while patients with severe iron overload may experience terminally irreversible heart failure symptoms [35]. Early identification of IOC is clinically important. Multiple physiologic, biomolecular and morphological factors such as tachycardia, hemodynamic overload, LV hypertrophy, endocrinopathies, genetic disposition, neuro-hormonal activation and pro-inflammatory cytokines could aggravate cardiac dysfunction [36-38]. In the early stages of the disease, ventricular systolic function remain well preserved and patients are asymptomatic. As the disease progresses, iron deposition occurs in the ventricular myocardium. Patients may present with exertional dyspnea due to left ventricular (LV) systolic dysfunction in the setting of restrictive pathophysiology [35,39,40].
Iron deposition then occurs in the atrial myocardium causing atrioventricular (AV) block and supraventricular arrhythmias, which correlate with the extent of iron deposition in atrial tissues [41]. Iron deposition in the conduction system may occur giving rise to nodal disease, which may cause bradycardias and increase the need for pacemaker placement [42]. The condition may progress into dilated cardiomyopathy characterized by LV dilation and paroxysmal atrial fibrillation leading to myocardial damage and increased risk of sudden cardiac death (SCD) in severely iron-overloaded patients [35]. Ventricular dilation and systolic dysfunction predisposes patients to more frequent ventricular arrhythmias. Right HF could also be present in the early stages of the disease, which may be independent of left HF or evolve and progress along with left HF [41,43]. Severe cardiac impairment reduces survival to one year [43]. Iron deposition may also occur in the pericardium, which in most cases is asymptomatic [5].
Despite the existence of different forms of IOC (primary and secondary), they share the same pathogenic mechanisms after significant levels of iron deposition has occurred in cardiac, hepatic and/or endocrine organs. In primary or HH-induced IOC, mutations in the genes encoding crucial proteins involved in iron-metabolism give rise to high duodenal iron absorption relative to the total body iron content leading to excess iron and its deposition on cardiomyocytes [24]. On the other hand, in secondary IOC, serum iron overload is the result of high exogenous iron intake principally through chronic blood transfusion that saturate the reticuloendothelial system cells with iron, which may spill out to other parenchymal cells [44,45]. Other secondary but lesser causes of increased serum iron overload include increased intake of exogenous (dietary) iron. Figure 1 provides a schematic illustration of the pathophysiology of IOC beginning with serum iron overload leading to myocardial iron overload, and eventually causing myocardial dysfunction. The bold lines indicated well-established pathogenetic mechanisms for IOC. The dotted lines indicate not fully established pathogenetic mechanisms, but which have the potential to contribute to iron-overload induced cardiomyopathy. Finally, the double lines indicated an indirect pathologic effect of endocrinopathies, hepatopathy and immune deficiency on myocardial dysfunction in patients with iron overload.
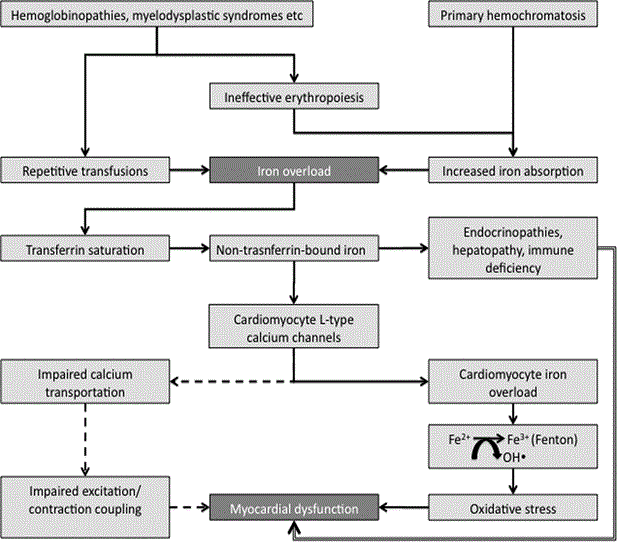
Figure 1: Pathogenic Mechanisms of Iron Overload Cardiomyopathy.
Iron overload cardiomyopathy occurs due to excess serum iron causing transferrin saturation, which releases non-transferrin bound iron into circulation. The non-transferrin bound iron enters cardiomyocytes via L-Type calcium channels and is catalyzed by Fenton reaction to produce reactive oxygen species that causes cell damage and death, eventually leading to cardiomyopathy. Adapted from Kremastinos & Farmakis, 2011, p. 2256 [14].
The deposition of iron in the myocardium is a gradual process, which depends on the degree of increase in the levels of serum iron. Under normal iron homeostasis (metabolism), transferrin-bound uptake mechanisms regulate cardiac iron. However, during iron overload, transferrin that is approximately 30% saturated becomes fully saturated and the toxic non-transferrin bound iron is released into circulation. Since the negative feedback mechanisms regulating transferrin-bound iron uptake does not control cellular uptake of non-transferrin-bound iron in concert with the lack of iron-excretory mechanism, leads to accumulation of intracellular iron. The uptake of iron from non-transferrin bound iron in hepatocytes, cardiac myocytes and endocrine glands cells causes iron to accumulate in tissues and consequent deleterious effects of iron overload [28]. During iron overload, ferrous iron (Fe2+) enters cardiomyocytes through the voltage dependent L-Type Calcium Channels (LTCC) [46,47]. Iron uptake in the myocardium is slower relative to hepatic uptake, which explains why myocardial iron overload develops much later compared to hepatic iron overload [48]. Pathological iron deposition occurs initially in the epicardium and then extends to the myocardium and finally to the endocardium. This progression helps to explain the preserved systolic function in the early stages of IOC [21].
Cardiomyocytes store iron in the form of ferritin, hemosiderin and labile cellular free iron, with labile being the most active. Labile iron is catalyzed by the rapid Fenton reaction, which converts ferrous to ferric ion with the generation of toxic hydroxyl ions, to form Reactive Oxygen Species (ROS) [49]. Upon exceeding anti-oxidant capacity of the cell, there is peroxidation of membrane lipids producing alterations in membrane permeability. These modifications produce a leak of hydrolytic enzymes initiating cell damage and subsequent myocyte death, eventually leading to the development of cardiomyopathy characterized by diastolic and systolic dysfunction [5,12]. Increased ferrous iron transportation through the LTCC could also cause alterations in cardiomyocytes calcium transportation and impaired excitation/contraction coupling, which may be involved in the development of diastolic and systolic dysfunction [20]. In cases of IOC occurring concomitant with myocardial ischemia (MI), iron overload may aggravate ischemia-associated reperfusion injury, which may result into an autocatalytic process and eventually in a cardiomyopathic process [21,51]. In addition to direct myocardial injury, iron overload may affect the heart indirectly through its effect on liver and endocrine glands. Hepatic dysfunction, endocrinopathies (diabetes mellitus, hypothyroidism, hypoparathyroidism), and immune deficiency resulting from iron overload may contribute to the pathophysiology of IOC [50,51].
The 2016 AHA Scientific Statement on Diagnostic and Treatment Strategies for specific forms of dilated cardiomyopathies suggests that a high index of clinical suspicion is key for the diagnosis of IOC. Morphological and functional features of IOC may mimic those of dilated cardiomyopathy and restrictive cardiomyopathy (RCM) and thus clinical suspicion of iron overload is important to suggest the need for additional tests to exclude DCM and RCM.
At present, there is no specific standardized guidelines or expert consensus on the diagnostic algorithm for IOC [19]. However, based on clinical experience with patients with cardiomyopathies, the National Institutes of Health Clinical Center Cardiology Consult Services proposed the following clinical pathway for the diagnosis of IOC (Figure 2).
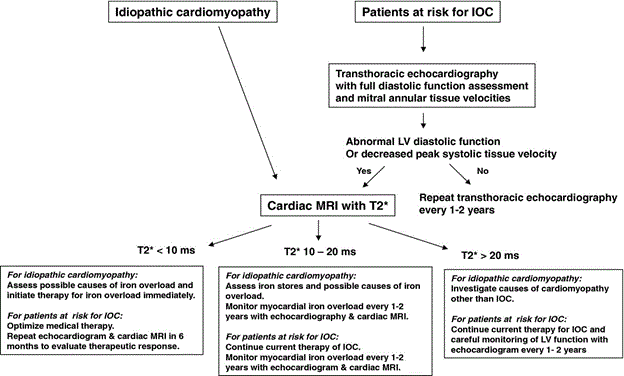
Figure 2: Diagnostic Pathway of Iron Overload Cardiomyopathy.
Serological tests (serum transferrin status and family history helps identify patients at risk (suspected) for IOC. Routine echocardiography helps to identify diastolic/systolic dysfunction. CMR T2 is the mainstay of diagnosis for IOC by quantifying myocardial and hepatic iron. Adapted from Gujja et al., 2010, p. 1007 [5].
The initial clinical evaluation for patients suspected with IOC or at risk of IOC is important to identify diagnostic clues for confirmation of IOC. Initial evaluation should include basic laboratory tests, patient history taking, physical examination, standard ECG and chest X-ray [14]. Upon clinical suspicion, the diagnosis of iron overload begins with basic laboratory investigation including serological tests and tests for end-organ involvement [19]. Initial laboratory investigation should target transferrin saturation (serum iron/total iron binding capacity > 45%) and serum ferritin (elevated > 200 µg/L in males or >150 µg/L in females) suggesting HH. Assessment of family history may reveal a known or suspected condition causing iron overload such as hemoglobinopathies, MDS, or other transfusion-dependent hereditary or acquired anemias or HH, suggesting the need for genetic testing to confirm HH genotype or hemoglobinopathies [24,53].
Physical examination is useful to reveal signs of left or right-sided HF and signs associated with iron-overload/injury such as typical skin pigmentation [14]. Standard ECG helps to identify supraventricular and ventricular arrhythmias as well as conduction abnormalities, which are part of IOC clinical phenotype [54,55]. Chest x-ray may reveal cardiomegaly due to LV enlargement in patients with dilated phenotype of IOC and clinical signs of pulmonary congestion in patients with left-sided heart failure, left atrial or RV enlargement in the presence or absence of signs of pulmonary hypertension in patients with restrictive IOC phenotype [14]. Additional tests may include full blood count, hemoglobin electrophoresis, liver function tests, endocrine tests (diabetes mellitus, thyroid and gonads) to evaluate underlying disorder causing iron overload and potential consequences of iron overload on organ function [44].
Transthoracic echocardiography (TTE) should be considered as part of initial and regular follow-up evaluation in patients suspected or at risk of IOC based on initial clinical evaluation or have had chronic RBC transfusions [19]. TTE should be done with complete LV diastolic function assessment including quantification of tissue velocity of the mitral annulus every 1 and 2 years irrespective cardiac symptoms or evidence of iron overload. TTE may detect alterations in LV/RV systolic and diastolic dysfunction and pericardial and valvular involvement. Diastolic LV dysfunction featuring pseudo-normalized or restrictive filling pattern in the presence or absence of atrial enlargement may constitute early TTE findings [56-62]. TTE is useful to detect advance-stage IOC marked with left and right cardiac chamber dilation and depressed LVEF (dilated phenotype_ or restrictive LV filling with left atrial and RV dilation increased pulmonary artery pressure and preserved LVEF (the restrictive phenotype) [61-63]. TTE could also detect high cardiac output with chamber dilation, eccentric LV hypertrophy and normal or increased LVEF [14,63].
Transthoracic echocardiography (TTE), although valuable in detecting consequences of iron on myocardial morphology and function and screening asymptomatic patients and follow-up of patients with known pathology, it does not reveal myocardial iron content [61,62,64]. According to 2016 AHA diagnostic recommendations, cardiac MRI T2* (star) relaxation time is currently the cornerstone of the diagnosis of IOC considered when TTE reveals abnormal LV diastolic function and/or depressed peak systolic tissue velocity of mitral annulus [19]. Introduced in 2001, Cardiac MRI T2* has revolutionized clinical management of patients with IOC and other iron overload conditions because it enables accurate diagnosis and quantification of myocardial and hepatic iron levels improving tailoring and monitoring of therapies and subsequent increase in survival rates of IOC patients [65].
Cardiac iron in the form of hemosiderin but not ferritin or labile affects T2* relaxation. However, because of a continuous reflux between the three forms of stored iron in the myocardium, cardiac MRI T2* is able to predict iron content in myocardial tissue [28,62]. In addition, cardiac MRI T2* assesses full-thickness area of interest in the inter-ventricular septum, which is highly representative of the global myocardial iron content [49]. The value of 20 ms is considered the threshold of myocardial siderosis [19]. Based on the threshold value, the National Institutes of Health Clinical Center Cardiology Consult Service [5,67] clinically grades the severity of IOC into three categories based on cardiac MRI T2* values as summarized in Table 3.
Table 3: Clinical Grading of the Severity of Iron Overload Cardiomyopathy
Cardiac MRI T2* Values |
Clinical Description |
T2* > 20 ms |
Green Zone: Patients at low risk for imminent development of congestive heart failure (CHF). Correspond to lack of iron overload or benign iron load associated with normal cardiac function with a high negative predictive value. |
10 ms ≤ T2* ≤ 20 ms |
Yellow Zone: Patients in whom cardiac deposition has probably occurred are at an intermediate risk of cardiac decompensation. Indicative of myocardial siderosis and an inverse correlation with LVEF. |
T2* < 10 ms |
Red Zone: Patients at the highest risk category of cardiac decompensation and most often require immediate review and intensification of therapy. Indicative of severe iron overload associated with an increased annual risk of the development of heart failure or arrhythmias |
The predictive value of cardiac MRI T2* 20 ms have been demonstrated to predict severe cardiac complications (especially heart failure) in patients with thalassemia major. Figure 3 shows the relationship between cardiac MRI T2* values (indicating cardiac tissue iron concentration) and the risk of heart failure in patients diagnosed with thalassemia major associated IOC. The risk of heart failure within a year increased from 0.2% (T2* = 10 to 20 ms) to 21.4% (T2* = 6 to 10 ms) and finally to 47.23% (T2* = 0 to 6 ms). Arrhythmias occurred in 19% (T2* < 6 ms), 18% (T2* 6 to 10 ms) and 4% (T2* > 10ms).
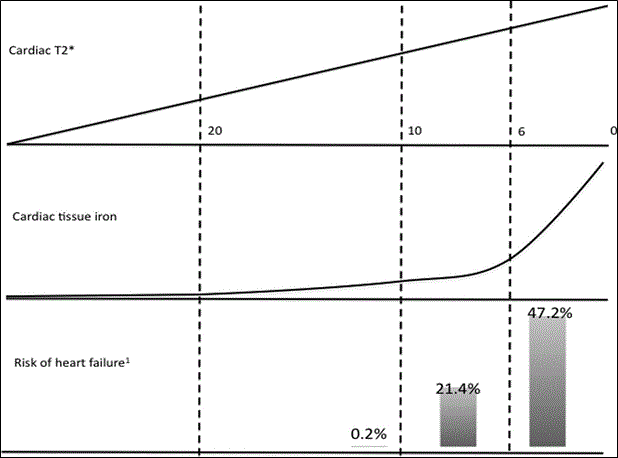
Figure 3: Relationship between T2* Values, and Iron Overload and the Risk of HF.
In a prospective study of 652 patients with thalassemia major, as cardiac MRI T2* values decreased from 20 ms to 10 ms and finally to 0 ms, the risk of heart failure increased significantly from 0.2% to 47.2%. Adapted from Kirk et al., 2009, p. [68].
Despite diagnostic accuracy of cardiac MRI T2*, its widespread implementation is severely limited by its restricted availability in developing nations. This restriction has influenced the use of the traditional tests of serum ferritin in affected developing nations. Serum ferritin increases linearly with the number of blood transfusions and closely correlates with hepatic iron content to predict iron overload [5]. It also provides a simple means of monitoring the efficacy of chelation therapy for IOC patients. However, serum ferritin is also an acute phase protein increasing in several other conditions and poorly correlates with myocardial iron load [69].
Confirmatory diagnosis of IOC is thus two-phase. The first phase should provide evidence of heart diseases, in particular diastolic LV dysfunction with restrictive filling or LV remodeling with chamber dilation and significantly depressed LV function (LVEF). The second phase should provide evidence of co-existence of iron overload (serum ferritin > 300 ng/mL; transferrin saturation > 55%; or cardiac MRI T2* > 20 ms) [44,64,65]. Besides identification of patients with established IOC, it is clinically important to identify patients at risk for developing IOC especially those with iron overload (with or without cardiac siderosis) and those with conditions potentially causing iron overload. Early identification is important since it improves prophylactic therapy against the development of IOC [14].
The mainstay of the diagnosis of IOC has targeted etiology-specific methods – to quantify myocardial iron. In this regard, Cardiac T2* MRI has emerged as a valuable diagnostic and prognostic tool for patients diagnosed with IOC based on its ability to quantify global myocardial and hepatic iron burden. In the case of unavailability of Cardiac T2* MRI, the assessment of LV function (defined as LVEF) and serum ferritin have also been used as important low-cost diagnostic methods. However, research evidence on the relationship between myocardial iron burden of Cardiac T2* MRI, and serum ferritin and LVEF have been inconclusive. The present meta-analysis combines research evidence on the correlation between Cardiac T2* MRI, and LVEF and serum ferritin values.
Study selection
Studies were searched from online databases: MEDLINE, EMBASE, CINAHL and clinical trials.gov. A broad combination of key words used to ensure all relevant studies are identified included “iron overload cardiomyopathy” OR “siderotic cardiomyopathy” OR “iron cardiotoxicity” AND “cardiac T2* MRI” OR “serum ferritin” OR “LV function”. Inclusion criteria were studies that: (a) recruited patients either prospectively or retrospectively; (b) recruited patients diagnosed iron overload cardiomyopathy or etiologic conditions; (c) used cardiac T2* MRI imaging to assess myocardial iron burden; and (c) reported main outcomes of diagnosis. Citation of descriptive or reviews, studies with only abstracts, studies that data was not readily extractable or case series (have limited sample) were excluded. However, there was no restriction on publication period or publication language. Extracted data comprised of first author, year of publication, research design, number of patients recruited, percentage of male patients, mean age, main etiology investigated, correlating diagnostic parameters and non-correlating parameters. Table 4 provides a summary of data abstracted from the included studies.
Table 4: Summary of Data on Diagnosis of Iron Overload Cardiomyopathy
1st Author [Ref.] |
Publication Year |
Study Sample |
Male (%) |
Mean Age (Yrs.) |
Main Etiology for IOC |
Global Myocardial Mean |
Correlating Diagnostic Parameters |
Not Correlating Diagnostic Parameters |
Anderson et al. [65] |
2001 |
106 |
60 |
27 |
Thalassemia Major |
< 20 ms normal 52±16 |
Reduced LVEF (r=0·61, P<0·001)
Increased in LVESV (r=0·50, P<0·0001 (r=0·40, P<0·001) |
Serum ferritin (r=0·10, p=0·32) |
Chacko et al. [70] |
2007 |
11 |
55 |
74 |
Transfusion-dependent MDS |
34 ms |
LVEF > 56% |
Serum Ferritin (r = -0.01; p = 0.96) |
Mavrogeni et al. [71] |
2005 |
25 |
|
|
β-Thalassemia |
35.7±3.7
42.9±2.4 |
LVEF = 44.1±2.8
42.9±2.4 |
Serum ferritin (r = -0.51, p = < 0.01) |
Mavrogeni et al. [72] |
2007 |
9 |
|
|
β-Thalassemia |
31.0±4.6 |
LVEF = 64.5±7 |
Serum ferritin liver |
Wood et al. [73] |
2004 |
19 |
53 |
16 |
Thalassemia Major |
26.1± 4.6 ms
Normal 35.7±5 ms |
LVEF 67±2
LVESV 29.1±2.8 |
Serum Ferritin (r=0.33, P=.01) |
Di Tucci et al. [74] |
2008 |
27 |
67 |
69 |
Acquired Anemia: Myelodysplastic Syndrome/ Primary Myelofibrosis |
Median 39.8 ms |
Transfusion burden (p = 0.0002) |
Serum ferritin (r = 0.25 (95%CI -0.17 to 0.59; p = 0.24) |
Roghi et al. [75] |
2010 |
49 |
59 |
41 |
Thalassemia Intermedia |
Mean = 38.7 ± 11.0 ms
Normal < 20 ms |
LVEF (p = 0.03) |
Serum ferritin (R2 = 0.003; p = 0.716) |
Konen et al. [76] |
2007 |
13 |
62 |
79 |
Myelodysplastic Syndrome/ Congenital Hemolytic Anemia |
< 20 ms |
LVEF Normal |
Serum Ferritin Liver |
Westwood et al. [77] |
2005 |
67 |
40 |
29 |
Thalassemia Major |
Normal < 20 ms |
Atrial Peak Filling Rate (APFR) r = 0.49, p < 0.001)
EPFR/APFR ratio (r = -0.62, p < 0.001) |
Early Peak Filling Rate (EPFR) (r = - 0.20, p = 0.19) |
MDS: Myelodysplastic Syndrome; APFR: Atrial Peak Filling Rate; EPFR: Early Peak Filling Rate
In total, nine (9) studies [65,70-77] meeting the inclusion criteria were included in the present meta-analysis. The nine studies investigated the correlation between Cardiac T2* MRI, and LVEF and serum ferritin in patients suspected with IOC. The combined population was 326 patients diagnosed with IOC or its main secondary transfusion-dependent etiologies. The mean age of the patients was 46.8 years, range 16 [73] to 79 years [76] with a slightly greater presentation of male patients (56.2%). The main cardiac condition associated with IOC investigated was transfusion dependent thalassemia [65,71-77] and Myelodysplastic Syndrome (MDS) [70,74]. In all the studies, a mean of cardiac T2* MRI was < 20 ms was the threshold of myocardial iron overload but normal values ranged from 52.0 ms [65] to 31.0 ms [72]. In all the nine studies, LVEF had a positive correlation with Cardiac T2* MRI defined myocardial iron burden. Myrocqdial iron burden < 20 ms correlated with lower values of LVEF. However, Cardiac T2* MRI myocardial iron values did not correlated with serum ferritin. Cardiac T2* MRI myocardial iron values also positively correlated with LVESV, atrial peak filling rate (APFR), Early Peak Filling Rate (EPFR) and EPFR/APFR ratio. However, Cardiac T2* MRI myocardial iron values had no correlation with serum ferritin values.
Iron deposition on the myocardium has been well established as the primary pathogenic mechanism of IOC. As such, the assessment of myocardial iron burden has been the cornerstone of clinical diagnosis of IOC [18,19]. The assessment of LV diastolic function or ventricular hemodynamics is also important for diagnosis of cardiomyopathy. In addition, the assessment of the levels of serum ferritin is an important low-cost method to quantify myocardial iron burden. However, the relationship between cardiac T2* MRI myocardial iron burden. The present analysis finds that: (a) Cardiac T2* MRI is able to quantify myocardial iron burden in IOC patients with a threshold of < 20 ms (normal); (b) Cardiac T2* MRI myocardial iron values has a significant correlation with LVEF function; and (c) Cardiac T2* MRI myocardial iron burden has no significant correlation with the levels of serum ferritin. Cardiac T2* MRI provides a rapid and reproducible technique for detecting myocardial iron burden equal to or greater than transfusion iron levels [74].
Consistent with previous studies, myocardial iron assessment using cardiac T2* MRI has considerable been enhanced in IOC patients. It has developed as a reference standard for myocardial iron characterization in transfusion-dependent patients in safely and reliably [61,62]. Normal cardiac T2* MRI values for myocardial iron (> 20 ms) correlated well with normal LV function (LVEF > 45%). However, the relationship between cardiac T2* MRI values for myocardial iron content did not have any relationship with serum ferritin and liver iron content. According to Westwood et al. [77], serum ferritin cannot predict myocardial iron content in the early stages of IOC but can only detect iron overload in patients with advanced disease. Although no correlation has been established between cardiac T2* MRI values and hepatic iron content, a long latent period relative to hepatic iron content predates the development of myocardial iron overload [70]. In patients with thalassemia intermediate, cardiac T2* MRI reveals no evidence of myocardial iron overload although hepatic iron accumulation is significant [75]. There is need for additional studies to determine when myocardial iron overload can occur in patients with thalassemia intermediate. Finally, although cardiac T2* MRI myocardial burden correlates well with diastolic parameters – EPFR, APFR and EPFR/APFR ratio, low sensitivity limits the use of diastolic parameters [77].
Currently there are no specific guidelines or expert consensus on the treatment of IOC. Current treatment protocols focus on treating the etiology of IOC. The cornerstone of clinical management of IOC is lowering serum iron levels in the body. The intention is to decrease the risk of developing cardiovascular abnormalities [19]. Although, current therapies for lowering serum iron focus on genetic (HH) and exogenous iron sources, phlebotomy and chelation therapy remain the mainstay of IOC treatment. Although dietary interventions may appear promising to minimize serum iron levels, it has negligible clinical effect. According to Hemochromatosis Management Working Group [70], approximately an extra 0.5 to 1.0 mg of ingested iron is absorbed daily in individuals with HH. Minimizing or eliminating this total daily dietary iron absorption thus has clinically insignificant effect in lowering serum iron levels. Moreover, diets do not enhance iron excretion and there is no substitute for iron depletion therapy [78]. However, patients may eliminate consumption of iron-rich food such as red meat and still have an effect on lowering total body iron levels. Particularly, alcohol increases iron absorption and its intake should be minimized [79]. Multi-vitamin tables containing iron and Vitamin C should also be avoided [80]. With limited value of dietary intervention on clinical management of IOC patients, the current proposed treatment algorithm focuses on chelation therapy and phlebotomy as schematically illustrated in Figure 4 for patients with established or suspected with IOC or patients at a greater risk of developing IOC.
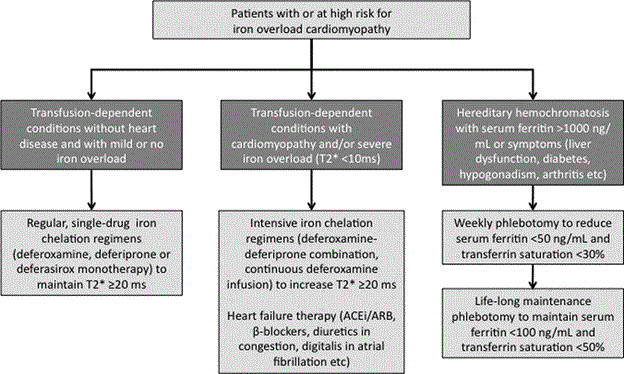
Figure 4: Clinical Management of Iron Overload Cardiomyopathy.
Current validated treatment of IOC targets the reduction of serum iron levels using chelation therapy for secondary IOC or phlebotomy for primary IOC and conventional medical therapy for symptomatic heart failure. Adapted from Kremastinos & Farmakis, 2011, p. 2260 [14].
Therapeutic phlebotomy
Therapeutic phlebotomy is the gold standard for treating HH. It causes iatrogenic anemia by withdrawing 400 to 500 ml of blood (about 200 to 250 mg of iron) at each session, leading to the body mobilizing iron from the organs where iron is stored for the production of hemoglobin [5]. It is indicated for HH patients when serum ferritin > 1000 ng/mL or in the presence of symptoms. In the introduction phase, phlebotomy may be done once or twice weekly to reduce serum ferritin < 50 ng/mL and transferrin saturation < 30% [24,81,82]. Upon achieving therapeutic ferritin levels, the frequency of the maintenance phlebotomy should be determined by periodic follow-up, with gradual reducing of phlebotomy intervals. Maintenance usually requires three to four phlebotomy for males and once or twice for females [83,84]. When initiated early, therapeutic phlebotomy can prevents the development of iron-induced organ damage, improvement in cardiac function and prolongs survival. However, phlebotomy cannot reverse established iron-overload associated complications such as liver cirrhosis and insulin-dependent DM as well as refractory arrhythmias have been noted in patients undergoing phlebotomy [85-87].
Chelation therapy is the medical treatment for metal poisoning recommended for the treatment of secondary iron overload in IOC patients (mainly with hemoglobinopathies and thalassemia major) without overt heart failure and contraindicated for phlebotomy because of significant anemia or malignancy and those exhibiting hemodynamic instability [5,19]. The therapeutic goal of chelation therapy is to detoxify organs with excess iron deposition by binding irons, removing them and excreting the compound in urine or bile [5]. The first potent iron chelator - parenteral deferoxamine, hexadentate chelator – was introduced in the 1970s. Other recently identified chelating agents are oral Deferasirox and Deferiprone, but their clinical use is limited because of ongoing research [44].
Parenteral deferoxamine is clinically approved and highly specific iron-chelating molecule, which works by binding iron released from reticuloendothelial system. It has a high affinity to bind with trivalent ferric ion and removes cardia iron by direct interaction. Chronic use of deferoxamine has clinically beneficial outcomes including decreased cardiac complications and increased survival in transfusion-dependent, iron-overloaded thalassemia patients [88-90]. High intravenous doses rather than the traditional subcutaneous infusion have also been used for rapid removal of cardiac iron from patients with heavy iron-overload and cardiac failure [91].
Deferiprone, a recently identified bidentate chelating agent with good oral bioavailability and rapid absorption in the stomach, has been investigated in patients with thalassemia and sickle cell with transfusion iron overload, but its chronic efficacy and safety have not been fully established [84]. Another recently identified chelator is deferasirox, a tridentate-lipophilic oral chelating agent. Although early clinical benefits have been demonstrated in thalassemia, sickle cell patients, myoproliferative disorders and Diamond-Blackfan anemia, results from several small-scale randomized clinical trials (RCTs) on the use of deferasirox have not been consistent [5]. Additional studies are warranted to demonstrate the value of chronic use of Deferasirox in the reduction of iron levels and the effect on cardiac function.
For transfusion dependent IOC patients with cardiac T2* MRI < 10 ms and cardiomyopathy or severe symptomatic heart failure and/or objective evidence of morphological or structural ventricular dysfunction, a dual therapy of intensified chelation therapy and conventional heart failure medication may be recommended [14]. If an IOC patients has reached New York Heart Association (NYHA) stage IV functional class heart failure symptoms and refractory heart failure symptoms despite aggressive chelation therapy, heart failure therapy including medication, cardiac resynchronization therapy and heart transplantation may be considered [5]. Conventional HF medical therapy including angiotensin converting enzyme (ACE) inhibitor/ Angiotensin receptor blockers (ARB) could be considered. Other cardioprotective medication may be used as appropriate in individual cases such as diuretics for relief of congestive heart failure and anticoagulation for atrial fibrillation and thromboembolism [14]. In IOC cases, presenting with acute HF, intravenous loop diuretics, inotropes, renal replacement therapy and circulatory support may be considered as per clinical needs of the patients [50].
Chelation therapy has dramatically prolonged life expectancy in patients with transfusion dependent secondary IOC, primarily Thalassemia major. However, the precise clinical benefit in reducing myocardial iron burden and improving cardiac function has not been fully established. Thus, the present systematic review and meta-analysis combines findings from clinical trials investigating chelation therapy on patients with iron overload cardiomyopathy to assess for reductions in iron burden and improvement in LV function, defined using LV ejection fraction.
The present systematic review and meta-analysis combines the findings of available published peer reviewed articles assessing the clinical efficacy of iron chelating agents on reducing myocardial iron burden and improving LV function (defined hereinafter as LVEF). Relevant studies were searched from online databases: MEDLINE, EMBASE, CINAHL and clinical trials.gov. A combination of key words included in the search were iron overload or burden, myocardial, iron cardiomyopathy, clinical trial, and chelation therapy. Inclusion criteria was studies that: (a) were clinical trials; (b) recruited patients diagnosed with secondary IOC – transfusion-dependent Thalassemia: used cardiac T2* MRI imaging to assess myocardial iron burden; and (c) reported outcomes of therapy. Citation of descriptive or reviews, studies with only abstracts and studies that data was not readily extractable were excluded. There was no restriction on publication period or publication language. Data was abstracted from each included studies. The extracted data comprised of first author, year of publication, research design, study sample (population), percentage of female patients, mean age of recruited sample, main etiology investigated, iron chelating agent used and its mean dose, unit of cardiac T2* MRI assessment at baseline and at follow-up and follow-up period. Two independent investigators resolved any discrepancy. Table 5 provides a summary of data abstracted from the included studies.
Table 5: Summary of Data on Chelation Therapy for Iron Overload Cardiomyopathy
1st Author [Ref.] |
Publication Year |
Research Design |
Study Sample |
Female (%) |
Mean Age (Yrs.) |
Chelating Agent |
Mean Dose (mg/kg/day) |
> T2* (ms) |
LVEF Change |
Follow-up (Months) |
Piga et al. [92] |
2014 |
Prospective Multicenter |
20 |
55 |
31 |
Deferasirox |
30.7 |
4.70 |
1.70 |
52 |
Wood et al. [93] |
2010 |
Prospective Multicenter |
28 |
70 |
23 |
Deferasirox |
33.3 |
3.40 |
1.30 |
18 |
Maggio et al. [94] |
2002 |
Retrospective Multicenter |
144 |
51 |
21 |
Deferasirox |
50.0 |
1.14 |
0.00 |
12 |
Deferiprone |
75.0 |
0.12 |
1.00 |
Pennell et al. [95] |
2006 |
Prospective Multicenter |
61 |
49 |
26 |
Deferoxamine |
43.0 |
2.10 |
0.30 |
12 |
Deferiprone |
92.0 |
3.50 |
3.1 |
Peng et al. [96] |
2003 |
Prospective |
24 |
|
|
Deferoxamine |
50.0 |
0.01 |
1.30 |
36 |
Deferiprone |
75.0 |
0.20 |
6.60 |
Tanner et al. [97] |
2007 |
Prospective |
65 |
|
|
Deferoxamine |
45.0 |
3.30 |
0.60 |
12 |
Deferiprone |
75.0 |
6.00 |
2.60 |
Kolnagou et al. [98] |
2006 |
Prospective |
11 |
36 |
|
Deferoxamine |
50.0 |
4.70 |
-- |
28 |
In total, 365 citations were retrieved from online search. After screening title, abstract and full article of the qualifying studies, seven studies investigating the effect of iron chelating agents on myocardial iron burden and LV function were included in this meta-analysis [92-98]. The seven studies recruited a combined sample of 353 patients with transfusion dependent thalassemia major with mean age 25 years, range 21 [94] to 31 [92] obtained from four studies [92-95]. Each gender was equally presented with female patients accounting for 52%. The seven studies investigated three iron chelators – deferasirox, deferiprone and deferoxamine. Three studies compared the efficacy of deferiprone and deferoxamine [95-97], while two studies also investigated the effect of dual therapy of deferiprone and deferoxamine [97,98]. The patients were followed up for a mean period of 24 months, range 12 [94-97] to 52 [92] months.
The seven studies had important differences in terms of location, patient population, duration of follow-up and measurement of cardiac iron using MRI. However, in general, early institution of iron chelation therapy on transfusion dependent thalassemia patients with significant iron overload (< 20 ms) with preserved ejection fraction cause a significant reduction in mean myocardial iron burden of 2.7 ms, range 0.2 to 6.0 ms to normal values (> 20 ms). In the four studies [94-97], comparing deferiprone and deferoxamine, there was no significant differences in the redsuction of myocardial iron burden. Assessing the effect of iron chelation on LV function, the findings reveal a marginal improvement in LV function in all the studies with relative increase in mean LVEF of 1.9% to the baseline values. However, the improvement was not statistically significant. The findings suggest iron chelation therapy does produce clinically significant changes in the LV function for patients in the early stages of IOC. However, the present meta-analysis included patients with significant iron overload but with preserved ejection fraction. Additional research on IOC patients with depressed LV function should provide evidence on the chronic effect of iron chelation on LV function.
The introduction of iron chelation therapy in the 1970s has made possible the management of patients with secondary iron overload who are intolerant to or contraindicated for phlebotomy [14]. Unlike phlebotomy that works by withdrawing blood from patients to minimize myocardial iron burden, chelation therapy uses iron-binding agents to enable excretion of the iron molecule [89]. Despite the value of chelation therapy in improving the survival and quality of life in transfusion-dependent thalassemia patients, the choice of an appropriate iron-chelating agent has remained a clinical challenge. Three key findings of the present meta-analysis is: (a) the use of iron chelation therapy conveys a distinct and significant clinical benefit by reducing myocardial iron burden. (b) The effect of iron chelation therapy on cardiac function is not clinically significant. (c) There is no significant differences in the clinical efficacy (reduction of myocardial iron burden) of the three currently available iron-chelating agents: deferasirox, deferiprone and deferoxamine.
The present findings indicate that iron chelation therapy achieves clinically significant reduction of myocardial iron content but does not cause any significant improvement on LV function. This could be explained by the criteria used for recruiting patients in the included studies. Most of the studies recruited patients with iron overload and preserved LVEF. Little changes may be expected from patients with normal LV function. Moreover, the studies reported mean LVEF at baseline and did not provide data for a subset of patients who had significantly depressed LV function at presentation and any changes after receiving chelation therapy. Although changes in LVEF were not significant, it could still imply an improvement in LV function. This is because transfusion-dependent thalassemia patients who are not under chelation therapy usually exhibit a progressive reduction in LV function [30,31]. Since there was no notable reduction in LVEF, the findings suggest improvement in cardiac function.
The present findings indicate iron-chelating agents have similar effect on reducing myocardial iron content. This finding fails to demonstrate an earlier study by Piga et al. [98] reporting deferiprone conveys a superior cardio-protection against iron cardiotoxicity than subcutaneous deferoxamine based on the reduction of myocardial iron content. The present findings further suggest that other than clinical efficacy in reducing myocardial iron burden, the choice of iron-chelating agents could depend on other important factors such as cost, intolerance or compliance. However, there is support for the continued use of deferoxamine as the first-line drug of choice for patients diagnosed with thalassemia major [94]. Although not investigated in the present meta-analysis, research support for dual therapy of iron chelating agents on reducing myocardial iron burden is accumulating. The use of a dual therapy of deferiprone and deferoxamine has been shown to have superior cardio-protection against iron overload than a single therapy of either of the two iron chelating agents [96-100]. However, it remains inconclusive on the cut-off value of serum ferritin or cardiac T2* MRI to initial a dual therapy [102].
The most serious and life-threatening complication of excessive serum iron is a cardiac disorder termed as iron-overload cardiomyopathy (IOC). The disorder weakens contractility of cardiac muscles and compromises the efficiency of blood circulation. It is an important and potentially reversible cause of heart failure involving diastolic dysfunction, increased susceptibility to arrhythmias and late-stage dilated cardiomyopathy hence its classification as an etiology or sub-type of dilated cardiomyopathy. IOC has two phenotypic expression – dilated phenotype involving LV remodeling causing chamber dilation and depressed LV function and restrictive phenotype involving diastolic dysfunction with restrictive filling, preserved LV function and subsequent RV dilation. Etiologies of IOC include primary causes due to genetic disorders of iron metabolism principally hereditary haemochromatosis and secondary causes due to excessive or chronic administration of exogenous iron through repeated blood transfusion or dietary sources. Clinically, at the early stages, patients are asymptomatic with preserved ventricular systolic function but as the disorder progresses, patients may present with exertional dyspnea due to developing LV dysfunction and eventually exhibit signs of heart failure. The main pathogenic mechanism of IOC is fully saturated transferrin. It results in the release of non-transferrin bound iron into circulation, which enters cardiomyocytes and is catalyzed by the rapid Fenton reaction to produce toxic hydroxyl ions to form reactive oxygen species causing alterations in membrane permeability leading to cell damage and necrosis, and eventually leads to myocardial dysfunction. Diagnosis of IOC is two phase. First, to provide evidence of diastolic LV dysfunction with restrictive filling or LV remodeling with chamber dilation and depressed LVEF using echocardiography. Second, to provide evidence of co-existing iron overload principally using cardiac T2* MRI, or serum ferritin concentration or transferrin saturation. The mainstay of clinical management of IOC is the reduction of serum iron through therapeutic phlebotomy for primary IOC mainly due to hereditary hemochromatosis, and chelation therapy for secondary IOC due to excessive or chronic exogenous iron administration.
References
- Coffey R, Ganz T2 (2017) Iron homeostasis: An anthropocentric perspective. J Biol Chem 292: 12727-12734. [Crossref]
- Gozzelino R, Arosio P (2016) Iron homeostasis in health and disease. International Journal of Molecular Sciences 17: 120-130.
- Muñoz M, García-Erce JA, Remacha ÁF (2011) Disorders of iron metabolism. Part II: iron deficiency and iron overload. J Clin Pathol 64: 287-296. [Crossref]
- Muñoz M, García-Erce JA, Remacha AF (2011) Disorders of iron metabolism. Part 1: molecular basis of iron homoeostasis. J Clin Pathol 64: 281-286. [Crossref]
- Gujja P, Rosing DR, Tripodi DJ, Shizukuda Y (2010) Iron overload cardiomyopathy: Better understanding of an increasing disorder. Journal of the American College of Cardiology 56: 1001-1012.
- Hall M (1830) Researches principally relative to the morbid and curative effects of loss of blood. Philadelphia: E.L. Carey and A. Hart.
- Gregory G, Broussais FJV (1830) Elements of the theory and practice of physic: designed for the use of students. Published by M. Sherman.
- Sullivan JL (1981) Iron and the sex difference in heart disease risk. Lancet 1: 1293-1294. [Crossref]
- Sullivan JL (1989) The iron paradigm of ischemic heart disease. Am Heart J 117: 1177-1188. [Crossref]
- Sullivan JL (1992) Stored iron and ischemic heart disease. Empirical support for a new paradigm. Circulation 86: 1036-1037. [Crossref]
- Sullivan,JL (1996) Iron versus cholesterol-perspectives on the iron and heart disease debate. Journal of clinical epidemiology 49: 1345-1352.
- Sullivan JL (1999) Iron therapy and cardiovascular disease. Kidney Int Suppl 69: S135-137. [Crossref]
- Das SK, Patel VB, Basu R, Wang W, DesAulniers, J, et al. (2017) Females are protected from iron-overload cardiomyopathy independent of iron metabolism: Key role of oxidative stress. Journal of the American Heart Association 6: e003456.
- Kremastinos DT, Farmakis D (2011) Iron overload cardiomyopathy in clinical practice. Circulation 124: 2253-2263. [Crossref]
- Das SK, Zhabyeyev P, Basu R, Patel VB, Dyck JR, et al. (2017) Advanced iron-overload cardiomyopathy in a genetic murine model is rescued by resveratrol therapy. Bioscience Reports
- Oudit GY, Backx PH (2016) Amlodipine therapy for iron-overload cardiomyopathy: The enduring value of translational research. Canadian Journal of Cardiology 32: 938-940.
- Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, et al. (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. European Heart Journal 37: 1850-1858.
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, et al. (2006) Contemporary definitions and classification of the cardiomyopathies. Circulation 113: 1807-1816.
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646.
- Murphy CJ, Oudit GY (2010) Iron-overload cardiomyopathy: pathophysiology, diagnosis, and treatment. Journal of Cardiac Failure 16: 888-900.
- Liu P, Olivieri N (1994) Iron overload cardiomyopathies: new insights into an old disease. Cardiovasc Drugs Ther 8: 101-110. [Crossref]
- Aydinok Y, Porter JB, Piga A, Elalfy M, El Beshlawy A, et al. (2015) Prevalence and distribution of iron overload in patients with transfusion-dependent anemias differs across geographic regions: results from the CORDELIA study. European Journal of Hematology 95: 244-253.
- Tantiworawit A, Tapanya S, Phrommintikul A, Saekho S, Rattarittamrong E, et al. (2016) Prevalence and risk factors for cardiac iron overload and cardiovascular complications among patients with thalassemia in Northern Thailand. Southeast Asian Journal of Tropical Medicine and Public Health 47: 1335-1342.
- Pietrangelo A (2004) Hereditary hemochromatosis--a new look at an old disease. N Engl J Med 350: 2383-2397. [Crossref]
- Camaschella C (2005) Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood 106: 3710-3717.
2021 Copyright OAT. All rights reserv
- Beutler E, Hoffbrand AV, Cook JD (2003) Iron deficiency and overload. Hematology Am Soc Hematol Educ Program. [Crossref]
- Silverstein SB, Rodgers GM (2004) Parenteral iron therapy options. Am J Hematol 76: 74-78. [Crossref]
- Wood JC (2009) History and current impact of cardiac magnetic resonance imaging on the management of iron overload. Circulation 120: 1937-1939. [Crossref]
- Weatherall DJ, Clegg JB (1996) Thalassemia--a global public health problem. Nat Med 2: 847-849. [Crossref]
- Weatherall DJ, Clegg JB (2001) Inherited haemoglobin disorders: an increasing global health problem. Bulletin of the World Health Organization 79: 704-712.
- Quinn CT, Rogers ZR, Buchanan GR (2004) Survival of children with sickle cell disease. Blood 103: 4023-4027. [Crossref]
- Gordeuk V, Mukiibi J, Hasstedt SJ, Samowitz W, Edwards CQ, et al. (1992) Iron overload in Africa. Interaction between a gene and dietary iron content. N Engl J Med 326: 95-100. [Crossref]
- Moyo VM, Mandishona E, Hasstedt SJ, Gangaidzo IT, Gomo ZA, et al. (1998) Evidence of genetic transmission in African iron overload. Blood 91: 1076-1082. [Crossref]
- McNamara L, MacPhail AP, Gordeuk VR, Hasstedt SJ, Rouault T (1998) Is there a link between African iron overload and the described mutations of the hereditary haemochromatosis gene? British journal of Hematology 102: 1176-1178.
- Kamran SH, Saleem U, Ahmad B, Ahmad M (2012) Effect of iron overload cardiomyopathy in haemochromatosis and ß-thalassemia. Journal of Applied Pharmacy 21: 556-566.
- Hahalis G, Alexopoulos D, Kremastinos DT, Zoumbos NC (2005) Heart failure in beta-thalassemia syndromes: a decade of progress. Am J Med 118: 957-967. [Crossref]
- Hahalis G, Manolis AS, Apostolopoulos D, Alexopoulos D, Vagenakis AG, et al. (2002) Right ventricular cardiomyopathy in ß-thalassaemia major. European Heart Journal 23: 147-156.
- Hahalis G, Manolis AS, Gerasimidou I, Alexopoulos D, Sitafidis G, et al. (2001) Right ventricular diastolic function in ß-thalassemia major: echocardiographic and clinical correlates. American Heart Journal 141: 428-434.
- Skinner C, Kenmure AC (1973) Haemochromatosis presenting as congestive cardiomyopathy and responding to venesection. British Heart Journal 35: 400-466.
- Wasserman AJ, Richardson DW, Baird CL, Wyso EM (1962) Cardiac hemochromatosis simulating constrictive pericarditis. The American Journal of Medicine 32: 316-323.
- Buja LM, Roberts WC (1971) Iron in the heart. Etiology and clinical significance. Am J Med 51: 209-221. [Crossref]
- Schwartz KA, Li Z, Schwartz DE, Cooper TG, Braselton WE (2002) Earliest cardiac toxicity induced by iron overload selectively inhibits electrical conduction. Journal of Applied Physiology 93: 746-751.
- Fitchett DH, Coltart DJ, Littler WA, Leyland MJ, Trueman T, et al. (1980) Cardiac involvement in secondary haemochromatosis: a catheter biopsy study and analysis of myocardium. Cardiovascular Research 14: 719-724.
- Brittenham GM (2011) Iron-chelating therapy for transfusional iron overload. N Engl J Med 364: 146-156. [Crossref]
- Ghoti H, Amer J, Winder A, Rachmilewitz E, Fibach E (2007) Oxidative stress in red blood cells, platelets and polymorphonuclear leukocytes from patients with myelodysplastic syndrome. European Journal of Hematology 79: 463-467.
- Tsushima RG, Wickenden AD, Bouchard RA, Oudit GY, Liu PP, et al. (1999) Modulation of iron uptake in heart by L-type Ca2+ channel modifiers: possible implications in iron overload. Circulation Research 84: 1302-1309.
- Wood JC (2008) Cardiac iron across different transfusion-dependent diseases. Blood reviews 22: S14-S21.
- Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, et al. (2003) L-type Ca 2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nature Medicine 9: 1180-1187.
- Carpenter JP, He T, Kirk P, Roughton M, Anderson LJ, et al. (2011) On T2* Magnetic Resonance and Cardiac Iron. Clinical Perspective. Circulation 123: 1519-1528.
- Aessopos A, Berdoukas V, Tsironi M (2008) The heart in transfusion dependent homozygous thalassaemia today–prediction, prevention and management. European Journal of Hematology 80: 93-106.
- Farmakis D, Giakoumis A, Aessopos A, Polymeropoulos E (2003) Pathogenetic aspects of immune deficiency associated with ß thalassemia. Medical Science Monitor 9: RA19-RA22.
- Horwitz LD, Rosenthal EA (1999) Iron-mediated cardiovascular injury. Vasc Med 4: 93-99. [Crossref]
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS (2011) Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54: 328-343.
- Jessup M, Manno CS (1998) Diagnosis and management of iron-induced heart disease in Cooley's anemia. Ann N Y Acad Sci 850: 242-250. [Crossref]
- Aessopos A, Farmakis D, Karagiorga M, Voskaridou E, Loutradi A, et al. (2001) Cardiac involvement in thalassemia intermedia: a multicenter study. Blood 97: 3411-3416.
- Spirito P, Lupi G, Melevendi C, Vecchio C (1990) Restrictive diastolic abnormalities identified by Doppler echocardiography in patients with thalassemia major. Circulation 82: 88-94.
- Kremastinos DT, Tsiapras DP, Tsetsos GA, Rentoukas EI, Vretou HP, et al. (1993) Left ventricular diastolic Doppler characteristics in beta-thalassemia major. Circulation 88: 1127-1135.
- Kremastinos DT, Toutouzas PK, Vyssoulis GP, Venetis CA, Vretou HP, et al. (1985) Global and segmental left ventricular function in beta-thalassemia. Cardiology 72: 129-139. [Crossref]
- Bosi G, Crepaz R, Gamberini MR, Fortini M, Scarcia S, et al. (2003) Left ventricular remodeling and systolic and diastolic function in young adults with ß thalassaemia major: a Doppler echocardiographic assessment and correlation with haematological data. Heart 89: 762-766.
- Kremastinos DT, Tsiapras DP, Tsetsos GA, Rentoukas EI, Vretou HP, et al. (1993) Left ventricular diastolic Doppler characteristics in beta-thalassemia major. Circulation 88: 1127-1135.
- Aessopos A, Farmakis D, Deftereos S, Tsironi M, Tassiopoulos S, et al. (2005) Thalassemia heart disease: A comparative evaluation of thalassemia major and thalassemia intermedia. Chest 127: 1523-1530.
- Aessopos A, Farmakis D, Hatziliami A, Fragodimitri C, Karabatsos F, et al. (2004) Cardiac status in well-treated patients with thalassemia major. European Journal of Hematology 73: 359-366.
- Kremastinos DT, Tiniakos G, Theodorakis GN, Katritsis DG, Toutouzas PK (1995) Myocarditis in beta-thalassemia major. A cause of heart failure. Circulation 91: 66-71. [Crossref]
- Kremastinos DT, Farmakis D, Aessopos A, Hahalis G, Hamodraka E, et al. (2010) ß-thalassemia cardiomyopathy: History, present considerations, and future perspectives. Circulation: Heart Failure 3: 451-458.
- Anderson LJ, Holden S, Davis B, Prescott E, Charrier CC, et al. (2001) Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. European Heart Journal 22: 2171-2179.
- Modell B, Khan M, Darlison M, Westwood MA, Ingram D, et al. (2008) Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. Journal of Cardiovascular Magnetic Resonance 10: 40-42.
- Wood JC (2007) Magnetic resonance imaging measurement of iron overload. Curr Opin Hematol 14: 183-190. [Crossref]
- Kirk P, Roughton M, Porter JB, Walker JM, Tanner MA, et al. (2009) Cardiac T2* magnetic resonance for prediction of cardiac complications in thalassemia major. Journal of Cardiovascular Magnetic Resonance 11: 1-10.
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS (2011) Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54: 328-343.
- Chacko J, Pennell DJ, Tanner MA, Hamblin TJ, Wonke B, et al. (2007) Myocardial iron loading by magnetic resonance imaging T2* in good prognostic myelodysplastic syndrome patients on long-term blood transfusions. British Journal of Haematology 138: 587-593.
- Mavrogeni SI, Markussis V, Kaklamanis L, Tsiapras D, Paraskevaidis I, et al. (2005) A comparison of magnetic resonance imaging and cardiac biopsy in the evaluation of heart iron overload in patients with ß-thalassemia major. European Journal of Haematology 75: 241-247.
- Mavrogeni S, Gotsis ED, Berdousi E, Ladis V, Verganelakis D, et al. (2007) Myocardial and hepatic T2* magnetic resonance evaluation in ex-thalassemic patients after bone-marrow transplantation. The International Journal of Cardiovascular Imaging 23: 739-745.
- Wood JC, Tyszka JM, Carson S, Nelson MD, Coates TD (2004) Myocardial iron loading in transfusion-dependent thalassemia and sickle cell disease. Blood 103: 1934-1936.
- Di Tucci AA, Matta G, Deplano S, Gabbas A, Depau C, et al. (2008) Myocardial iron overload assessment by T2* magnetic resonance imaging in adult transfusion dependent patients with acquired anemias. Haematologica 93: 1385-1388.
- Roghi A, Cappellini MD, Wood JC, Musallam KM, Patrizia P, et al. (2010) Absence of cardiac siderosis despite hepatic iron overload in Italian patients with thalassemia intermedia: An MRI T2* study. Annals of Hematology 89: 585-589.
- Konen E, Ghoti H, Goitein O, Winder A, Kushnir T, et al. (2007) No evidence for myocardial iron overload in multitransfused patients with myelodysplastic syndrome using cardiac magnetic resonance T2* technique. American Journal of Hematology 82: 1013-1016.
- Westwood MA, Wonke B, Maceira AM, Prescott E, Walker JM, et al. (2005) Left ventricular diastolic function compared with T2* cardiovascular magnetic resonance for early detection of myocardial iron overload in thalassemia major. Journal of Magnetic Resonance Imaging 22: 229-233.
- Barton JC, McDonnell SM, Adams PC, Brissot P, Powell LW, et al. (1998) Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med 129: 932-939. [Crossref]
- Conrad ME, Barton JC (1980) Anemia and iron kinetics in alcoholism. Semin Hematol 17: 149-163. [Crossref]
- Felitti VJ, Beutler E (1999) New developments in hereditary hemochromatosis. Am J Med Sci 318: 257-268. [Crossref]
- Porter JB (2001) Practical management of iron overload. Br J Haematol 115: 239-252. [Crossref]
- Tung BY, Kowdley KV (1998) Clinical management of iron overload. Gastroenterol Clin North Am 27: 637-654. [Crossref]
- Andrews NC (1999) Disorders of iron metabolism. N Engl J Med 341: 1986-1995. [Crossref]
- Bring P, Partovi N, Ford JAE, Yoshida EM (2008) Iron overload disorders: treatment options for patients refractory to or intolerant of phlebotomy. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 28: 331-342.
- Candell J, Lu L, Seres L, Gonzalez JB, Batlle J, et al. (1983) Cardiac hemochromatosis: beneficial effects of iron removal therapy: an echocardiographic study. The American Journal of Cardiology 52: 824-829.
- Rivers J, Garrahy P, Robinson W, Murphy A (1987) Reversible cardiac dysfunction in hemochromatosis. American Heart Journal 113: 216-217.
- Dabestani A, Child JS, Henze E, Perloff JK, Schon H (1984) Primary hemochromatosis: anatomic and physiologic characteristics of the cardiac ventricles and their response to phlebotomy. American Journal of Cardiology 54: 153-159.
- Aletras AH, Ingkanisorn WP, Mancini C, Arai AE (2005) DENSE with SENSE. J Magn Reson 176: 99-106. [Crossref]
- Zurlo MG, De Stefano P, Borgna-Pignatti C, Di Palma A, Piga A, et al. (1989) Survival and causes of death in thalassaemia major. Lancet 2: 27-30. [Crossref]
- Gabutti V, Piga A (1996) Results of long-term iron-chelating therapy. Acta Haematol 95: 26-36. [Crossref]
- Marcus RE, Davies S, Bantock HM, Underwood SR, Walton S, et al. (1984) Desferrioxamine to improve cardiac function in iron-overloaded patients with thalassemia major. The Lancet 323: 392-393.
- Piga A, Longo F, Origa R, Roggero S, Pinna F, et al. (2014) Deferasirox for cardiac siderosis in ß-thalassaemia major: A multicentre, open label, prospective study. British Journal of Hematology 167: 423-426.
- Wood JC, Kang BP, Thompson A, Giardina P, Harmatz P, et al. (2010) The effect of deferasirox on cardiac iron in thalassemia major: impact of total body iron stores. Blood 116: 537-543. [Crossref]
- Maggio A, d'Amico G, Morabito A, Capra M, Ciaccio C (2002) Deferiprone versus deferoxamine in patients with thalassemia major: a randomized clinical trial. Blood Cells, Molecules, and Diseases 28: 196-208.
- Pennell DJ, Berdoukas V, Karagiorga M, Ladis V, Piga A, et al. (2006) Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood 107: 3738-3744.
- Peng CT, Chow KC, Chen JH, Chiang YP, Lin TY, et al. (2003) Safety monitoring of cardiac and hepatic systems in ß-thalassemia patients with chelating treatment in Taiwan. European Journal of Haematology 70: 392-397.
- Tanner MA, Galanello R, Dessi C, Smith GC, Westwood MA, et al. (2007) A randomized, placebo-controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation 115: 1876-1884.
- Kolnagou A, Kontoghiorghes GJ (2006) Effective combination therapy of deferiprone and deferoxamine for the rapid clearance of excess cardiac IRON and the prevention of heart disease in thalassemia. The Protocol of the International Committee on Oral Chelators. Hemoglobin 30: 239-249.
- Piga A, Gaglioti C, Fogliacco E, Tricta F (2003) Comparative effects of deferiprone and deferoxamine on survival and cardiac disease in patients with thalassemia major: a retrospective analysis. Haematologica 88: 489-496.
- Daar S, Pathare AV (2006) Combined therapy with desferrioxamine and deferiprone in beta thalassemia major patients with transfusional iron overload. Ann Hematol 85: 315-319. [Crossref]
- Kattamis A, Ladis V, Berdousi H, Kelekis NL, Alexopoulou E, et al. (2006) Iron chelation treatment with combined therapy with deferiprone and deferioxamine: a 12-month trial. Blood Cells Mol Dis 36: 21-25. [Crossref]
- Hershko C, Link G, Konijn AM, Cabantchik ZI (2005) Objectives and mechanism of iron chelation therapy. Ann N Y Acad Sci 1054: 124-135. [Crossref]