Keywords
hypermobility, connective tissue dysplasia, ehlers-danlos syndrome, beighton scale, dysautonomia, postural orthostatic tachycardia syndrome, irritable bowel syndrome, mast cell activation disorder, articulo-autonomic dysplasia
Introduction
Vastly under-recognized and tragically mistreated are the 20% of women and 10% of men whose tissues are more flexible and fragile than average [1]. Few opportunities for prevention and treatment exceed that of recognizing hypermobility, easily screened by asking the patient to push their thumb against their forearm (Figure 1). This tell-tale sign can point the way to a more detailed evaluation that is well within the realm of the general practitioner, their broad clinical knowledge nicely matching the multisystem problems that may arise from lax connective tissue. Here I review the nature of hypermobility disorders, demonstrate that general rather than subspecialty knowledge can lead to life-changing therapies for these patients, and show how genetic testing brings symptomatic descriptions like fibromyalgia or chronic fatigue syndrome into the discerning light of precision medicine.
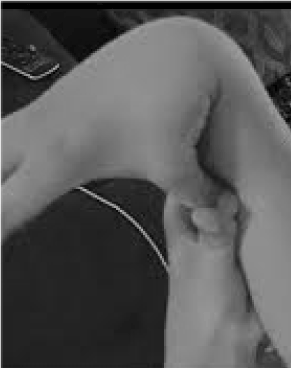
Figure 1. Hypermobility.
Several barriers must be overcome before hypermobility patients can garner the benefits of general medicine. The first derives involves the pioneering but myopic focus of dermatologists Ehlers and Danlos [2] imprinting incredible and exotic skin stretching as the misleading symbol of connective tissue laxity disorders. A second barrier is the increasing divide between practical and genetic medicine, the former appropriately concentrating on common disease while the latter until recently using specialized technology to describe the rare. The obscure eponyms with daunting subtypes and technical terms are like small provinces of an uninviting foreign country that no global traveler will take time to map. Countering these obstacles is a practical approach to hypermobility disorders that focuses on recognition by history and physical, leaving the technicalities of eponyms, subtypes, and genetic testing to subspecialists contacted after the vital first step of presumptive diagnosis. Once recognized and referred the flexible patient remains dependent on their primary physician, for only they can integrate recommendations and testing results into the general medical home.
Terminology
Increased range of motion at multiple articulation (joint) sites occurs in a large number of diseases, ranging from chromosome disorders like Down syndrome to disorders like Ehlers-Danlos syndrome (EDS) known for their hypermobility. McKusick [3] popularized this category of disorders known for tissue laxity by grouping them as heritable disorders of connective tissue, including Marfan syndrome that he focused on as a cardiologist. In 1977, Dr. Peter Beighton, known for his scale measuring flexibility, organized the intervening literature by postulating 6-7 EDS types, now simplified by recent guidelines as EDS classical, vascular, and hypermobile [4]. It is now clear that these types have considerable overlap of symptoms among each other and related disorders like osteogenesis imperfect (with numerous fractures), Marfan syndrome (large vessel dilation/dissection), Stickler syndrome (eye and arthritis changes), and many others.
Rather than be concerned with particular diseases, I find it more useful to focus on two major disease processes from connective tissue dysplasias, illustrated by the many symptoms listed in Table 1. First is the wear-and-tear joint pain (osteoarthritis) that differs from the swollen, red joints of inflammatory (autoimmune or inflammatory) arthritis encountered in rheumatoid arthritis, lupus, Sjogren syndrome, or ankylosing spondylitis. Second is the autonomic nervous system imbalance (dysautonomia) caused by elastic blood vessels in hypermobility disorders [5]. Distensible vessels cause pooling of blood in the pelvis and legs, with lower blood pressure and dizziness/faintness on standing (orthostatic hypotension). Over time, decreased blood flow to the head activates the sympathetic arm of the autonomic nervous system (the adrenalin “flight or fight” arm), leading to several problems: 1) postural orthostatic tachycardia syndrome (POTS) with tachycardia, anxiety, chronic fatigue, and “brain fog” (intervals of decreased focus and memory), 2) irritable bowel syndrome due to inhibition of intestinal motility (parasympathetic suppression) with constipation alternating with diarrhea, bloating, reflux, stomach pain, dysphagia, nausea, and 3) mast cell activation disorder with transient rashes, hives/reactive skin, asthma/shortness of breath, and food/medication intolerances. I have devised a broader term that facilitates recognition of any hypermobility disorder, recognizing the two main consequences of joint/tissue laxity: articulo-autonomic dysplasia or joint-fatigue complex.
Table 1. Typical findings and their frequency in hypermobility syndromes. *From 320 female and 54 male patients over age 12 years assessed by detailed questionnaires in 2017; TMJ, temporomandibular joint; POTS, postural orthostatic tachycardia syndrome. Symptom list adapted in part from Wilson GN, Cooley WC: Preventive Health Care for Children with Genetic Condition: Providing a Medical Home. 2nd ed. Cambridge, MA. Cambridge University Press, 2006.
HISTORY (percentages)* |
Infancy: Colic (28), feeding difficulties (19.4); clumsy, many falls (29.1) |
Child/teen: Slow weight gain (12); aware of flexibility (55.6); showed off joint tricks (53.6)
early joint pain (42.1); teen activities limited by pain-injury (55.4); early glasses (46.8)
orthodontics (61.4%) gum disease (11.3); poor enamel/many cavities (31.6) |
Joint issues: Popping (81.6); subluxuxations (62); TMJ popping, catching, pain (51.2)
pain in at least 2 joints (80.3); joint surgeries (33); many sprains (26); fractures >2 (31.5)
spinal disc herniation (29.3) |
Skeletal issues: Scoliosis (31); pectus (2.2); gait-toeing in/out (9.3); flat feet (15.5) |
Skin: Easy bruising (61); stretchy skin (15); velvety (18); unusual scars (36.3)
striae before pregnancy (73); slow healing (32.4) |
Genitourinary (females): Menorrhagia (61.2); endometriosis (11.3); ovarian cysts (55)
polycystic ovarian syndrome (8.5); bladder issues (46.1); inguinal/femoral hernia (11.5) |
Neuromuscular: Migraines (51.7); daily headaches (62.3); needed headache medicine (45)
Chiari deformation (10.6); neurosurgery (2.9); numbness/tingling (48.7); poor balance (48.6)
neuropathic shooting pains (37.3); muscle aches/spasms (51.7); muscle weakness (32.4) |
GI/bowels: Constipation-diarrhea (67.1); bloating-reflux-stomach pain (57.3)
gall bladder issues (12.9); difficult swallow (23); frequent nausea (44.9) |
Heart: Mitral valve prolapse (8.7); aneurysm (0.9); arrhythmia other than tachycardia (6.8) |
POTS: Dizzy on standing (69.8); syncope (34.5); chronic fatigue (81.8); sleep difficulties (43.5)
brain fog (poor memory or focus at times—70.8); heat/cold sensitivity (77.5); abnormal sweating (25); tachycardia (72.8); anxiety-panic attacks (61.3); salt fancy (51.2) |
Immune/mast cell: Transient rashes (33.4%); hives/reactive skin (45.9%); asthma/shortness of breath (39.3); food-medication intolerances (61.5%) |
Lab/Imaging: Low bone density (2.1); low vitamin D (28.7); low vitamin B12 (2.7); hypothyroid (11.5); low ferritin (1.7); low iron (6.7); elevated anti-nuclear antibody (7.1) |
Average number findings out of 80: females >12 (35.4 findings) males >12 (21.5 findings) |
Physical (percentage) |
Build: Tall-height >90th percentile (55.2); heavy-BMI >28 (21.3); thin-BMI <19 (14.2)
2021 Copyright OAT. All rights reserv
Marfanoid habitus (44); arachnodactyly (46.4); Walker-Murdoch sign present (40.2) |
Face: Long (25.7); tight lower face (5.4); blue-gray sclera (1.6); high palate (53.5) |
Skin: Soft (81.4); translucent (23.8); elastic-lift >1 inch fold around jaw on mid-forearm (51.3)
Folds consist of outer (epidermal) layer (35.4); unusual scars (42.1%) |
Beighton score: 0-3 points (8.4); 4-6 points (32.6); 7-8 points (38.8); 9 of 9 points (20.2) |
Other maneuvers: Join hands over shoulder-behind back (68)
upward prayer sign behind back (71.3); bring hand around back to touch umbilicus (19.5) |
Skeletal: Neck kyphosis (41.2); scoliosis (22.3); lordosis (32.8); flat feet (39.6)
gait-toeing in or out (28.3) |
Neurologic: Muscle weakness (11.2); decreased muscle mass (8.8)
poor balance by tandem walk (33.4) |
Average number findings out of 40: females >12 (17.9 findings) males >12 (15.7 findings) |
Articulo-Autonomic Dysplasia (AAD)
A term like “articulo-autonomic dysplasia” or AAD can unify the approach to connective tissue laxity/hypermobility disorders and counter three problems that complicate their recognition: 1) many physicians and geneticists view connective tissue dysplasias as rare and extreme diseases when in fact they affect about 1% of the population 2) EDS diagnostic guidelines [4], though simplifying major EDS types as classical, vascular, and hypermobility, still confuse by referring to many minor types that form an overlapping symptom spectrum, and 3) the almost obligate dysautonomia that accompanies EDS and related disorders (Table 1) is omitted as a diagnostic criterion, analogous to describing an elephant without mentioning its trunk. AAD denotes the inevitable physiologic connective between loose tissue and dysautonomia in the same way that the metabolic or hepatorenal syndromes describe how obesity leads to liver disease/diabetes or liver failure causes kidney disease.
Like most medical patterns or syndromes, including EDS, AAD is a symptom complex with many underlying causes. “Articulo” refers to the joint, recognizing that affliction of any articular component—skin, nerve, muscle, bone, connective tissue, blood vessel —cause a similar pattern of joint-tissue laxity with autonomic imbalance. There will be skin fragility, decreased bone density with susceptibility to fractures, muscle weakness with pain (myalgia), neuropathy with numbness/tingling, carpal tunnel, or regional pain syndromes, autonomic changes including tachycardia, anxiety, non-restorative sleep, bowel immotility, hives/rashes, food-medication intolerances, all present to variable extent even within specific “types” of EDS. I have not found great differences among patients fitting particular types of EDS, illustrated by more frequent skin changes in type I classical cEDS versus greater joint flexibility in type III hypermobile hEDS. The frequencies of symptoms in 374 patients registered by systematic query did not vary significantly among those I diagnosed as cEDS versus hEDS, and even the 8 patients identified as possible vascular vEDS by having collagen type III gene mutations were not distinctive (aneurysms present in one of the 8 patients had the same low frequency in other EDS categories). Most important is the concept of an EDS spectrum, best represented in my opinion as AAD.
Recognition of the AAD symptom complex, caused by EDS and related connective tissue dysplasias, will prevent sad patient journeys from specialist to specialist, symptomatic diagnoses like vertigo, fainting, fibromyalgia, or chronic fatigue, and tragic suffering when many preventive strategies and treatments are available. Furthermore, recognition of dysautonomia explains the anxiety from POTS and the depression that frequently accompanies chronic pain or activity limitation [6], behavior differences that can be recognized as consequences of medical rather than psychiatric disease.
Genetic testing supports the articulo-autonomic dysplasia concept
Implied by their characterization as heritable disorders of connective tissue [3], the more extreme forms of EDS and related disorders were found to exhibit autosomal dominant inheritance, meaning that affected individuals have one normal and one abnormal form of the gene (allele). Dominant inheritance makes sense because defective protein produced by the abnormal allele, analogous to a warped brick, would mix with normal bricks from the other allele and make a wobbly structure. Recognition of the joint or articulation as a multi-component structure, constrained by overlying skin, supported by interior bone and cartilage, and acting through intrinsic nerve, muscle, tendon, joint lining, and vessel, in turn implies that any of these elements can be impacted to produce dysplasia, a tissue disruption distinct from the malformations caused by other genetic disorders. The development of DNA cloning and sequencing would confirm the operation of autosomal dominant inheritance and the ability of genes altering any element of joint structure to cause the symptom pattern of AAD.
Genetic testing began with a focus on a “candidate” gene selected because of patient symptoms, illustrated by fibrillin-1 gene testing for Marfan syndrome [7]. Sequencing the gene DNA AGCT letters, the “text” of nucleotides, could reveal substitutions or “typos” that altered the amino acid of the encoded protein and its intended function. Then came the new technology called NextGen or rapid parallel DNA sequencing, analogous to reading all pages of a book at once rather than from end to end [8]. This allowed sequencing of gene groups (panels) like those causing cardiomyopathy or connective tissue dysplasia or even all 23,000 genes in each human cell (our genome) in 2-3 months [9]. Sequencing the protein-coding or exonic gene regions, called whole exome sequencing (WES), can be ordered from many companies and in my practice was obtained for less than $200-300 out-of-pocket cost in half of my patients (list price is $9000 and the amount billed to insurance is around $20,000).
Although interpreting the average 12,000 DNA variants detailed by WES remains a problem, diminished by a growing epidemiology of variants in normal and disease populations, yields of gene changes contributing to AAD symptoms among 620 patients undergoing WES in my practice is over 60%. In addition to over 80 mutations collagen genes, including those in collagen type I usually associated with osteogenesis imperfecta, collagen type V often associated with classical EDS, collagens type VI, IX, and XII initially described with myopathies, and the feared collagen type III associated with vascular EDS, are recurring mutations in genes affecting skin, nerve, muscle, fibrillar, and vessel components that reinforce a concept of AAD where alteration of any joint component produces the sign and symptom pattern outlined in Table 1. Now recognition of EDS and related disorders encompassed by the AAD concept can buttress clinical impression with objective DNA evidence that allows counseling of patient and family.
Diagnosis and therapy of hypermobility disorders/AAD
Alertness for the main symptoms of joint pain/fragility and autonomic (bowel, heart, immune disruption) can lead to quick tests for hypermobility as shown in Figure 1. The thumb to forearm sign can preface the entire Beighton scale, giving one point bilaterally for fingers pressed back beyond 90 degrees on the hand, touching thumbs to forearms, hyperextending the elbow with arms extended, hyperextending knees to form a backward leg curve, and 1 point for the ability to touch palms to the floor [10]. A brief survey of symptoms like those in Table 1 [11,12] can confirm suspicion, aided perhaps by including in the physical examinations some of the assessments listed in the lower part of Table 1. Note that not all AAD patients are tall or thin, and that a substantial portion will be over-weight due to restrictions of activity from pain and fatigue. Many will have elastic skin, examined by pulling up folds around the jaw or on the forearm, particularly when these are the thin, superficial folds of epidermis. Build-up of white tissue or keloid formation around scars is common, and assessment of Beighton or other hypermobility maneuvers listed in Table 1 can be helpful. A slumped posture with forward neck curve, scoliosis, and lordosis may be noted along with flat feet or toeing-out (more than in) upon walking. Muscle strength will be down in some patients along with poor balance by tandem walk, but the generalist with limited time needs only attend to a few of these physical signs; referral will establish the rest.
Most important will be orthopedists familiar with the osteoarthritis and companion changes in EDS like degenerative joint changes, deteriorating cartilage, plica bands, and regions of dead bone. An experienced and conservative orthopedist who recognizes poor outcomes of surgeries to “tighten” or replace joints is desirable. A cardiologist can document POTS using the tilt-table test for orthostatic changes in heart rate and blood pressure, also performing electro- and echocardiographic studies to exclude aortic dilation or aneurysms. Neurology can assess common migraines or daily headaches, the Chiari deformation that occurs with slippage and herniation of lax brain tissue, the neuropathy due to constriction within lax joints, and the muscle changes denoted by the symptom description fibromyalgia and highlighted by frequent myopathic gene changes that is fostering a new term of myopathic EDS [4,13]. Allergists familiar with mast cell activation and gastroenterologists who can document low bowel motility are valuable additions to the subspecialty team, and no one is more valuable than the primary physician for managing the many recommendations and medications that may be required. Although it may seem odd coming from a geneticist, I think the subspecialty evaluations that lead to therapy rate higher than genetic evaluation and testing, but the latter can be helpful in defusing assumptions about psychiatric disease [14].
Treatment begans with an exercise program that can benefit both arthritis due to joint hypermobility and its reflexive dysautonomia. Attention to joint pain (growing pains occur at night) or injury/skeletal deformity can institute prevention by making the patient aware of their condition (note in Table 1 that many do not recognize their flexibility as abnormal). Wear-and-tear injury can be prevented by bracing susceptible joints, waist bands for back support, and choosing joint-protective activities like swimming over jogging. Patients should remain active with moderate weight-lifting and other reasonable activities to build muscles around the joints, preventing cycles of inactivity that exacerbate chronic fatigue or fibromyalgia. Appropriate physical therapy, pain management, exercise guidance, and anecdotal medications like glucosamine/chondroitin preparations have helped many, but relaxants that take away muscle protection of joints and steroid injections that mask injury, promote osteoporosis/degeneration, and give only temporary relief should be avoided.
The mainstay of dysautonomia therapy is hydration (8-10 glasses fluid per day), salt (absent labile hypertension), and a high protein diet, all aimed at increasing blood fluid volume and improving cerebral circulation. Additional nutritional therapy can include vitamin C (2 g per day) vitamin D (>2000 units per day, more than the 400-800 in multivitamins), vitamin B12 (1-2.5 mg per day). Low intestinal motility or gastroparesis is helped by avoiding fluids before meals, smaller and more frequent meals, probiotics, enzyme preparations and, by report of many, lower gluten and dairy that are common irritants. Supine exercise programs, biofeedback, and many medication options are available through cardiologists for POTS (e. g., beta-blockers, Midodrine, Florinef), through neurologists or pain management experts (e. g., Gabapentin, Lyrica), or through allergists who can provide antihistamine protocols to suppress mast cells (e.g., Zyrtec, Zantac, Cromolyn, Singulair). Advantages of genomic testing like WES that goes beyond genes known to cause connective tissue dysplasia are novel findings like the muscle-related genes now known to cause EDS and documentation of incidental or secondary findings like the breast-ovarian cancer gene mutations. Finding of genes associated with more severe symptoms allows patients to know risks for offspring and to choose prenatal or preimplantation genetic diagnosis strategies compatible with their values.
Case Presentation
Eve came to her doctor at age 18, dizzy, faint, anxious, and sick to her stomach, so exhausted that she retired Friday evenings to rest all weekend. She had been healthy, active in gymnastics and cheer, a brave and bright girl whose perseverance despite injury gained her coaches applause. Then influenza from which she never recovered, nausea so severe that she could not eat, weight loss, fainting in class, waking at night with a pounding heart, waking in morning without restorative sleep. Eve saw many doctors, even the ER when palpitations and panic took her breath away. Nothing wrong except stress they said, all in her mind. Calmed but not cured by pills, work failing from fatigue and confusion, she wondered if she was going mad. Unrecognized was her soft and fragile skin, her hard and heavy periods, her daily headaches, and, most striking of all, hypermobility that she and her family assumed was normal: “Yes, we’re bendy and double-jointed, isn’t everyone?” Eve would have answered if asked.
Fortunately Eve sought a new primary physician who was able to document many of the signs and symptoms listed in Table I. Eve had early colic and reflux with difficulty breastfeeding, was very flexible, called the “boneless” baby by her mother, and when older would sit on her knees with her legs extended behind her (the “W”-position), saying it was more comfortable. She was initially slow to walk, falling down frequently and appearing clumsy, but soon was active and climbing everywhere, making her parents wince by putting a foot around her neck or doing the splits with ease. But as she entered grade school she began to complain of arthralgia, her legs hurting at night with her knees and ankles bothering her after activity. Her pediatrician reassured her parents that she was just having “growing pains,” but they became more concerned when she tried to play soccer and kept turning her ankles over, one sprain requiring a boot for 2 months after a stress fracture was noted.
Eve had to stop soccer but found she excelled at gymnastics as she grew older, choosing floor exercises because of poor balance on the beam and a tendency for her shoulder to pull out on the parallel bars. She noticed that her joints would pop with movement and often popped them on purpose because it seemed to relieve tension and pain. She tried out for cheerleading with success at maneuvers but found that she was too sensitive to heat. The other girls called her “tomato face” as she become overheated during routines, and she would tire easily, often needing a day of rest after exercise. Her digestive problems resurfaced with frequent constipation and stomach pain, often feeling bloated and needing laxatives. By the 6th grade she was missing school because of her bowel issues and joint pain, often feeling fatigued and dizzy. She didn’t feel rested in the morning and would often take naps on the weekends, a departure from her earlier active and enthusiastic lifestyle. She often felt anxious, not able to trace it to any specific school or family stress, and began to have headaches in the morning she thought were due to her poor sleep. Her parents again took her to her pediatrician and were again told there was nothing wrong but usual school stress.
When she was 14 Eve had a bad flu-like illness and missed school for a month with severe fatigue, headaches, nausea, vomiting, and weight loss. She had terrible dizziness and some chest pain, feeling her heart was pounding at times (having palpitations). Her parents sought another pediatrician’s advice and she sent Eve to a cardiologist, who thought she was having tachycardia from stress and anxiety surrounding her illness. He gave Eve propranolol, a beta-blocker, to slow her heart rate, but she became even more fatigued and was now bed-ridden much of the day. Her joint pain increased after her illness and she would linger in bed most of the day, feeling sad and becoming anxious when her parents tried to bring her outside or take her for a drive. They brought her back to the second pediatrician who referred her to psychiatry, and Zoloft was begun to help with anxiety and depression. After few days Eve broke out in a rash with hives and had to stop that medication. She continued to have dizziness and was seen by two more specialists, a gastroenterologist who thought she might have celiac disease and an ear-nose-throat specialist who diagnosed vertigo and sent her to neurology. She told that doctor about headaches emanating from the back of her head, and a supine head MRI study was obtained that was normal.
Appreciation of the likely AAD diagnosis by her primary physician instituted a different round of subspecialty referrals including neurology where an upright head MRI study demonstrated the herniation of Chiari deformation. A cardiologist documented an abnormal tilt-table test along with low bowel motility and proved that her anxiety, and stress-fatigue were due to dysautonomia. Nutritional hydration-salt-high protein strategies with beta-blocker and antihistamine therapy improved her fatigue and bowel symptoms and lessened her headaches. It was decided the Chiari herniation of 4-5 mm was not sufficient to require surgery, and her psychiatric management was recognized as treating secondary symptoms of a medical disorder. Some of her subspecialists, skeptical of her clinical diagnosis, were convinced by genetic testing that documented a change in a gene for the alpha-1 chain of type V collagen, abbreviated COL5A1, a common finding in patients with EDS. Eve had inherited the mutation from her mother, who had had milder AAD symptoms and was now able to manage them better.
Over several months Eve’s health and vitality were restored and she “got her life back,” as she and her parents would say. She still had joint pain, anxiety, and fatigue on occasion, but “wise exercise” to build muscle protection around her joints, nutritional approaches for dysautonomia, and biofeedback approaches to head off anxiety were life-changing [15-17]. These treatments were all because of recognition by her family practitioner, now able to coordinate care for Eve and her mother and to provide resources for understanding both EDS [18] and its underlying gene changes [19]. Transitioning to the next generation was the ability to inform Eve of her 50% risk to pass on her COL5A mutation and some risks for early labor and postpartum bleeding in any future pregnancy [20].
References
- Remvig L, Jensen DV, Ward RC (2007) Epidemiology of general joint hypermobility and basis for the proposed criteria for benign joint hypermobility syndrome: review of the literature. J Rheumatol 34: 804-809.
- Steinman B, Royce PM, Superti-Furga A (1993) The Ehlers-Danlos syndrome. In: Connective Tissue and its Heritable Disorders. B Steinman, PM Royce (Eds) Wiley-Liss: New York pp. 351-407.
- McKusick VA (1956) Heritable disorders of connective tissue. CV Mosby: St. Louis.
- Bloom L, Byers P, Francomano C, Tinkle B, Malfait F (2017) The International Consortium on the Ehlers-Danlos Syndromes. Am J Med Genet Part C Semin Med Genet 175: 5–7.
- Gazit Y1, Nahir AM, Grahame R, Jacob G (2003) Dysautonomia in the joint hypermobility syndrome. Am J Med 115: 33-40. [Crossref]
- Pizzo PA1 (2013) Lessons in pain relief--a personal postgraduate experience. N Engl J Med 369: 1092-1093. [Crossref]
- Pyeritz RE (2008) A small molecule for a large disease. N Engl J Med 358: 2829-2831. [Crossref]
- 8. Wyandt HE, Wilson GN, Tonk VS. Chapter 11: Gene and genome sequencing: Interpreting genetic variation at the nucleotide level. In: Human Chromosome Variation: Heteromorphism, Polymorphism, and Pathogenesis, Ed.2. Springer Nature, Singapore
- Mefford HC (2012) Diagnostic exome sequencing--are we there yet? N Engl J Med 367: 1951-1953. [Crossref]
- http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers–Danlos syndrome, hypermobility type. Am J Med Genet A 12: 2989-3004.
- Simpson MR (2006) Benign joint hypermobility syndrome: evaluation, diagnosis, and management. J Am Osteopath Assoc 106: 531–536.
- Kirk JA, Ansell BM, Bywaters EG (1967) The hypermobility syndrome. Musculoskeletal complaints associated with generalized joint hypermobility. Ann Rheum Dis 26: 419-425.
- Wilson GN (2014) Presymptomatic and preimplantation genetic diagnosis: Neurology, NextGenetics, and the next generation. JAMA Neurol 71: 403-404.
- Castori M, Morlino S, Celletti C (2012) Management of pain and fatigue in the joint hypermobility syndrome (a.k.a. Ehlers- Danlos syndrome, hypermobility type): principles and proposal for a multidisciplinary approach. Am J Med Genet A 158: 2055-2070.
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers–Danlos syndrome, hypermobility type. Am J Med Genet A 161A: 2989-3004.
- Bathen T, Hangmann AB, Hoff M (2013) Multidisciplinary treatment of disability in Ehlers-Danlos syndrome hypermobility type/ hypermobility syndrome: a pilot study using a combination of physical and cognitivebehavioral therapy on 12 women. Am J Med Genet A 161A: 3005-3011.
- EDS (2018) Ehlers-Danlos Syndrome National Foundation.
- OMIM (2018) Online Mendelian Inheritance in Man.
- Wilson GN (2014) Exome analysis of connective tissue dysplasia: death and rebirth of clinical genetics? Am J Med Genet A 164A: 1209-1212. [Crossref]