Introduction
Non-alcoholic fatty liver disease (NAFLD) is a growing public health concern in the United States (US). Two-thirds of the US population are overweight or obese, and approximately 80% of obese patients have NAFLD [1]. The prevalence of NAFLD was estimated at 31% in 2011-2012 [2] and continue to rise following the rising trend in obesity. A large Veterans’ Affairs study revealed that the prevalence of NAFLD more than doubled from 80 to 161 per 100,000 from 2001 to 2013 [3].
NAFLD is a continuum of disease ranging from benign hepatic steatosis to non-alcoholic steatohepatitis (NASH) to fibrosis and cirrhosis. Not all hepatic steatosis will progress to NASH or beyond. Studies have demonstrated that only 43-44% of patients with steatosis progressed to NASH, and 7-30% of NASH patients progress to cirrhosis [1,4–7]. The overall risk of hepatic steatosis progressing to cirrhosis is estimated at 1-2% [8]. To date, the mechanism of progression in NAFLD is not well understood. This review aims to discuss several proposed mechanisms in development of NASH and NASH-related cirrhosis.
Mechanisms of progression
The most widely accepted theory in the pathogenesis of NAFLD is described by a two-hit model. The first “hit” is infiltration of hepatotoxic free fatty acids (FFAs) in the liver, making it vulnerable to a second hit. Hepatic lipotoxicity results from generation of toxic lipid metabolites from FFAs, such as ceramides, diacylglycerols, lysophosphatidyl choline, and oxidized cholesterol metabolites [2]. The metabolites act as reactive oxygen species (ROS), which impose oxidative stress on hepatocytes [3]. Oxidative stress mainly impacts the mitochondria and endoplasmic reticulum and contributes to hepatocyte dysfunction [9]. The second “hit” can be any number of insults ranging from impaired mitochondrial function, drug toxicity, iron overload, ischemia, disordered adipokine activity, or other inflammatory damage [2,8].
Gut microbiome is thought to have some role in the pathogenesis of NAFLD. The precise contribution is unclear, however there are several possible influences. First, the intestinal flora can alter bile acid metabolism, which can affect lipid handling and fatty deposition in the liver [10].
The intestinal flora can also produce metabolites and/or endotoxins, which may be hepatotoxic and provide the “second hit” in the two-hit model [10]. The microbiome also alters host immunity, which may contribute to inflammation [10]. Intestinal bacteria can release pathogen- associated molecular patterns (PAMPs) for damage- associated molecular patterns (DAMPs), which act on toll-like receptors (TLRs) to promote inflammatory response [11].
FFAs also interact with sonic hedgehog ligand (SHH), which plays a key role in hepatic regeneration by recruiting inflammatory mediators to the site to clear debris from damaged cells. SHH is released by ballooned degenerated hepatocytes as signal for cellular regeneration [12]. With fatty infiltration and hepatocyte damage, SHH is upregulated and activates hepatic stellate cells, sinusoidal endothelial cells, and other inflammatory cells [12]. These cells, in turn, release pro-inflammatory cytokines, and other fibrogenic mediators, which contribute to inflammation and fibrosis.
Inflammatory cytokines are crucial to the development of steatohepatitis. The current literature cites several specific cytokines that have a role, including TNF-a, IL-6, IL-1β, IL-17, and IL-18 [2,8,11]. JNK-1 is a kinase involved in signaling cascade for apoptosis and inflammatory response. In the presence of excess adipose tissue and insulin resistance, activation of the JNK-1 pathway increases the circulating levels of these inflammatory cytokines [2]. Additionally, fatty infiltration of the liver upregulates TGFβ. In the setting of inflammation, hepatic stellate cells become activated by TGFβ and secrete extracellular matrix, ultimately causing liver fibrosis [2].
Adiponectin is an adipokine with anti-inflammatory and anti-fibrogenic activity that protects liver parenchyma against steatosis and apoptosis [10]. In obese patients, adiponectin levels are decreased. A study in mice showed decreased hepatic adiponectin signaling in the livers of NASH mice compared to mice with benign hepatic steatosis [13]. Although similar studies have not yet been conducted in humans, the animal data suggests that lack of adiponectin plays a role in development of NASH.
Genetic, environmental and lifestyle factors are likely to have some influence on advancing fatty liver disease. However, these are not yet well studied. Certain genes including patatin-like phospholipase domain-containing 3(PNPLA3),transmembrane 6 superfamily 2 (TM6SF2),and glucokinase regulatory protein (GCKR) have been implicated as predisposing factors in progression of NAFLD to NASH [10,12].
Figure 1 depicts the histologic progression of NAFLD and summarizes the potential mechanism that contribute to advancing disease.
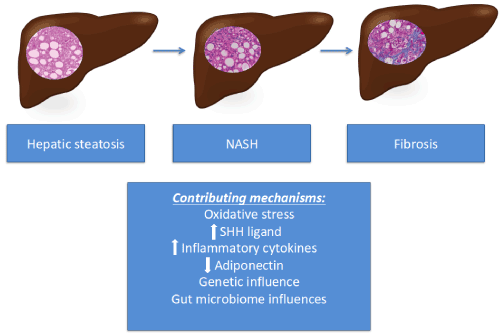
Figure 1 The progression of NAFLD and contributing mechanisms
Conclusion
With its rising prevalence, NAFLD is currently the second leading indication for liver transplant in the US and is expected to exceed the HCV as the leading cause soon. The key to reversing this trend is preventing progression of NAFLD to NASH and NASH cirrhosis. We must first have a clear understanding of the mechanisms underlying progression. Although there are many proposed mechanisms contributing to the advancement of NAFLD, further research is needed to understand how each of the mechanisms described above fit into the overall pathogenesis. This may also allow opportunities for novel interventions, such as use of antioxidants, anti-inflammatory agents, or agents that mediate adipokines, to halt advancement of disease.
References
- Satapathy SK, Sanyal AJ (2015) Epidemiology and Natural History of Nonalcoholic Fatty Liver Disease. Semin Liver Dis 35: 221-235. [Crossref]
- Peverill W, Powell LW2 Skoien R (2014) Evolving concepts in the pathogenesis of NASH: beyond steatosis and inflammation. Int J Mol Sci 15: 8591-8638. [Crossref]
- Beste LA, Leipertz SL, Green PK, Dominitz JA, Ross D, et al. (2015) Trends in burden of cirrhosis and hepatocellular carcinoma by underlying liver disease in US veterans, 2001-2013. Gastroenterology 149: 1471-1482. [Crossref]
- Golabi P, Sayiner M, Fazel Y, Koenig A, Henry L, et al. (2016) Current complications and challenge in nonalcoholic steatohepatitis screening and diagnosis. Expert Rev Gastroenterol Hepatol 10: 63-71. [Crossref]
- Traussnigg S, Kienbacher C, Halilbasic E, Rechling C, Kazemi-Shirazi L, et al. (2015) Challenges and Management of Liver Cirrhosis: Practical Issues in the Therapy of Patients with Cirrhosis due to NAFLD and NASH. Dig Dis 33: 598-607. [Crossref]
- Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, et al. (2015) Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systemic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol 13: 643-654. [Crossref]
- McPherson S, Hardy T, Henderson E, Burt AD, Day CP, et al. (2015) Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol 62: 1148-1155. [Crossref]
- Hassan K, Bhalla V, El Regal ME, A-Kader HH (2014) Nonalcoholic fatty liver disease: a comprehensive review of a growing epidemic. World J Gastro 20: 12081-12101. [Crossref]
- Ashraf NU, Sheikh TA (2015) Endoplasmic reticulum stress and oxidative stress in the pathogenesis of non-alcoholic fatty liver disease. Free Radic Res 49: 1405-1418. [Crossref]
- Caligiuri A, Gentilini A, Marra F (2016) Molecular Pathogenesis of NASH. Int J Mol Sci 17. [Crossref]
- Takaki A, Kawaku D, Yamamoto K (2014) Molecular mechanisms and new treatment strategies for non-alcoholic steatohepatitis (NASH). Int J Mol Sci 15: 7352-7379. [Crossref]
- Machado MV, Diehl AM (2016) Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 150: 1769-1777. [Crossref]
- Handa P, Maliken BD, Nelson JE, Morgan-Stevenson V, Messner DJ, et al. (2014) Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology 60: 133-145. [Crossref]
2021 Copyright OAT. All rights reserv