21-hydroxylase deficiency (21-HD) is a rare enzyme defect in the cortisol synthesis pathway. Left untreated, chronic elevations of adrenocorticotropic hormone induce adrenal cortex hyperplasia and may result in the formation of adrenal gland tumors. Benign myelolipomas and testicular adrenal rest tumors are commonly seen, but adrenocortical carcinomas (ACC) are extremely rare with a poor prognosis. We present the case of a 34-year old Caucasian man with untreated 21-HD, and a family history of liver, pancreatic and colon cancer, as well as 21-HD. Incidental computed tomography (CT) imaging for hip pain, and subsequent cancer work up, identified primary ACC with metastasis to the liver, lungs, bones, testicles and contralateral adrenal gland. After a precipitous decline in clinical status on the eighth day of admission, repeat CT imaging revealed worsening hepatomegaly and an interval enlarged left adrenal mass with active hemorrhage. The patient expired the following day due to rapid progression of aggressive metastatic cancer causing multi-organ failure. ACC is associated with hereditary cancer syndromes including Multiple Endocrine Neoplasia 1 (MEN-1), Lynch Syndrome, and Familial Adenomatous Polyposis (FAP). The association of MEN-1 with pancreatic cancer, and of Lynch and FAP with colon cancer, suggests the patient’s robust family history may have contributed to early tumorigenesis and rapid progression of disease in the setting of 21-HD. This case emphasizes the significance of compliance with steroid treatment in patients with 21-HD, as well as highlights the importance of early genetic testing when there is a concomitant family history of cancer.
21-hydroxylase deficiency, adrenocortical carcinoma
Congenital Adrenal Hyperplasia (CAH) is a disease characterized by a group of enzyme deficiencies in the production of mineralcorticoids, glucocorticoids, or sex steroids by the adrenal glands. Insufficient hormone production fails to induce negative feedback on upstream adrenocorticotropic hormone (ACTH) secretion, resulting in chronic elevations of ACTH. Long-standing stimulation of the adrenal cortex by ACTH results in adrenocortical hyperplasia and high levels of precursor hormones proximal to the enzyme defect. The most common enzyme deficiency, comprising 90% of CAH cases, is 21-hydroxylase deficiency (21-HD), which is characterized by low to normal levels of aldosterone and high levels of androgens. CAH is a rather uncommon disease, with an incidence of 1:1000 to 1:60,000 births in Caucasian populations [1].
CAH is classified into two main groups: classic CAH (CCAH) and non-classic (NCCAH) [1].
CCAH may be further divided into a simple virilizing form and a severe salt-wasting form [1]. The simple virilizing form occurs in approximately 25% of patients with 21-HD and is due to a point mutation that causes low but detectable enzyme levels. Aldosterone levels are normal, but there are still decreased levels of cortisol. Females are diagnosed at birth with ambiguous genitals, while boys are diagnosed later in childhood, after signs of androgen excess begin to display [1]. The salt-wasting form occurs in the remaining 75% of patients with 21-HD. In these patients, symptoms usually present earlier in life and are characterized by excessive salt wasting, precocious puberty in males, virilization in females at birth, and excessive hair growth.
In NCCAH, 21-hydroxylase activity is not as severely impaired as in CCAH, with reductions of only around 20% to 50% [1]. Thus, hyperandrogenism is present in NCCAH without salt wasting or genital malformations. The main symptoms are excessive hair growth and menstrual irregularities in women [2,3].
Treatment varies depending on the diagnosis of either CCAH or NCCAH. Pediatric cases of CCAH are treated with 10 to 15 mg/m2 of hydrocortisone divided into three doses daily [2,3]. Symptomatic NCCAH is treated with exogenous glucocorticoids to suppress ACTH levels and androgen production via induced negative feedback [2]. Asymptomatic NCCAH, on the other hand, is considered mild and only requires monitoring [2,3]. As CAH patients progress into adulthood, treatment becomes more difficult, as strict follow-up and adherence to medication regimens is required. Lack of continuous steroid treatment can result in adrenal cortex hyperplasia due to overstimulation from persistently high ACTH levels. The resulting adrenal cortex hyperplasia can further develop into benign tumors, or in rare cases, adrenocortical carcinoma (ACC) [4].
A 34-year-old Caucasian man, with a previous diagnosis of CAH, presented with bilateral hip and leg pain, and increasing dyspnea of one month duration. He visited his local emergency department (ED) several times where he was treated with non steroidal anti-inflammatory drugs (NSAIDs) and an oral dexamethasone taper for pain management. An abdominal computed tomography (CT) scan was taken on his third visit to the ED for worsening symptoms, revealing a 17 x 11 cm right adrenal mass with central necrosis and several smaller multifocal masses throughout the liver, lungs, left adrenal gland and spine. He was transferred to our hospital for cancer evaluation. On initial assessment, he was in mild distress from pain, which was exacerbated with any movement. Vital signs were normal except for oxygen saturation in the high 80s on room air. Physical exam was significant for diffuse abdominal tenderness and macroorchidic testes.
Further elucidation of his history revealed 21-hydroxylase deficiency diagnosed in early childhood, although he could not specify having the classic or nonclassic form. He also endorsed impotence, infertility, and abdominal cryptorchidism treated surgically with right sided orchidopexy in the neonatal period. He received daily glucocorticoid treatment until eighteen years of age when he elected to discontinue therapy and Endocrinology follow-up. He denied any hospitalizations due to adrenal insufficiency since withdrawal of steroid therapy. Of note, his family history was significant for liver and pancreatic cancer in his father, and colon cancer and 21-hydroxylase deficiency in his brother.
Objective data included a chest radiograph that showed bilateral hilar enlargement, suggestive of adenopathy, and right lung consolidation with a small right pleural effusion. A whole-body positron emissions tomography (PET)/CT scan obtained two days after admission re-demonstrated the masses seen on initial outside hospital CT and noted them to be hypermetabolic, consistent with widely metastatic cancer. CT-guided core needle biopsy of an easily accessible liver mass was performed on hospital day two, and pathology confirmed metastatic poorly differentiated adrenocortical carcinoma, staining positive for synaptophysin, alpha-inhibin, calretinin, and mismatch repair proteins MLH1, MSH2, MSH6, PMS2 (Figure 1). Ultrasound of macroorchidic testes also supported metastatic disease, as bilateral testicular and epididymal tissue were replaced with heterogeneous material.
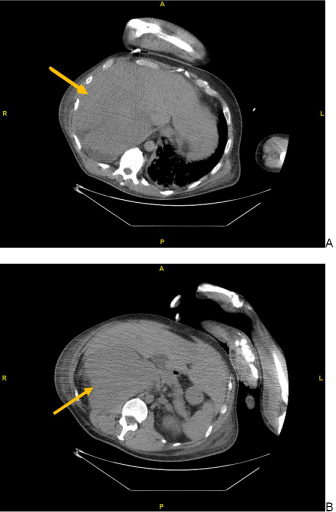
Figure 1. Abdominal CT images during biopsy of a liver mass on hospital day 2. (A) Hepatomegaly apparent with multi focal liver masses, and (B) a 17x11 cm right adrenal mass
In the setting of adrenal gland involvement and the patient’s history of 21-hydroxylase deficiency, Endocrinology was consulted and recommended obtaining relevant hormone levels (Table 1). A low-dose (1 mg) dexamethasone suppression test (LDDST) was performed, after which cortisol levels were found to be inappropriately normal. Post LDDST ACTH levels were not obtained due to the lag time in result reporting from a send-out lab. Once metastatic disease was established with imaging and pathology, Oncology was consulted and recommended obtaining various tumor markers which were significant only for a lactate dehydrogenase (LDH) of 2011 U/L (140-280 U/L). Serial routine labs were only notable for moderate increases in aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, total bilirubin and direct bilirubin, all concerning for progressive liver involvement.
Table 1. Serum and urinary hormone levels
Hormone |
Result |
RV for male population |
Expected* |
Observed |
ACTH |
15.5 |
0.0-46.0 pg/mL |
high |
normal |
Renin |
1.3 |
0.2-5.4 ng/mL |
high |
normal |
Aldosterone |
2.4 |
0.0-30.0 ng/dL |
low |
normal |
Cortisol, baseline |
22.45 |
4.3-23.0 ug/dL |
low |
normal |
Cortisol, post-LDDST |
20.17 |
4.3-23.0 ug/dL |
low |
normal |
17-OHP |
10850 |
27.0-199.0 ng/dL |
high |
high |
Androstenedione |
291 |
27.0-152.0 ng/dL |
high |
high |
Testosterone, Total |
140 |
241-827 ng/dL |
high |
low |
ACTH: adrenocorticotropic hormone
LDDST: low-dose dexamethasone suppression test
17-OHP:17-hydroxyprogesterone
RV: reference value
*Expected in an adult male with 21-HD
The patient’s subjective symptoms of pain and dyspnea were attributed to mass effect and remained stable throughout the majority of his hospitalization. His bilateral hip and leg pain were adequately controlled with NSAIDs and narcotics. He required continuous oxygen via nasal cannula at two liters per minute to maintain oxygen saturations at 89% to 95% at rest. Having achieved a stable clinical status and completed evaluation for cancer, following Oncology and Endocrinology recommendations, the patient was ready for discharge on oxygen, with plans to begin outpatient immunotherapy.
However, on hospital day eight, the patient rapidly decompensated, becoming acutely confused and severely hypotensive. He had an increased oxygen demand, new onset scleral icterus, abdominal distension, and bilateral pedal edema. Labs were consistent with acute liver failure. Repeat CT imaging showed worsening bilateral lung consolidative disease, worsening hepatomegaly, and an interval enlarged left adrenal mass with active hemorrhage (Figure 2). The patient was diagnosed with rapid progression of aggressive metastatic cancer and was transferred to the medical intensive care unit, where he was started on corticosteroids and supportive care. The following day, after an end-of-life-care discussion with his family, he was transitioned to comfort care and expired. His family declined an autopsy.
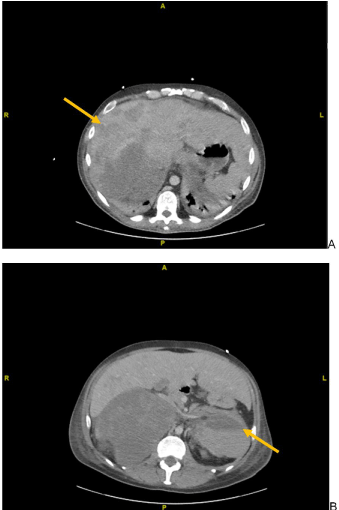
Figure 2. Abdominal CT images following clinical deterioration on hospital day 8. (A) Worsening hepatomegaly and enlargement of multi focal liver masses, and (B) enlarged left adrenal gland now measuring 11.8 x 8.3 cm with active contrast extravasation
Inadequate suppression of ACTH may lead to neoplastic transformation of adrenal cortical tissue in CAH [5]. Cessation of glucocorticoid therapy in patients with CAH may therefore result in long-standing ACTH elevations that induce adrenocortical hyperplasia and eventual tumorigenesis. Myelolipomas are considered the most common adrenal tumor in CAH patients, representing 1.5% to 9% of all adrenal incidentalomas [6]. They are benign and composed of mature adipose cells and hematopoietic tissue [7]. Chronic elevations in ACTH may also stimulate steroidogenic stem-cell-like tissue present in the testicles, resulting in testicular adrenal rest tumors (TARTs) in up to 94% of patients with CAH [8]. TARTs are well-defined, often bilateral, hypoechoic lesions of the testicle near the rete testes [9]. Although also typically benign, TARTs are commonly associated with impaired sperm production due to an increased proportion of nonfunctional testicular tissue. Surgical excision of myelolipomas and TARTs is reserved only for the presence of suspicious features of malignancy, such as heterogeneity or hemorrhage.
Although most adrenal masses and TARTs in the setting of CAH are benign and found incidentally, our patient presented with multiple alarm signs and pathology concerning for late metastatic malignancy. Our patient presented with a large right adrenal mass with central necrosis and hemorrhage, and with bilateral testicular masses of heterogenous material. His history of cryptorchidism repaired with orchidopexy instead of orchiectomy may have also contributed to testicular malignancy as orchidopexy does not reduce the risk of cancer [10]. Few cases of malignancy have been reported as a result of CAH, making this case of rapidly evolving ACC particularly alarming [11].
Additionally, our patient’s adrenal hormone levels during hospitalization were inconsistent with 21-hydroxylase deficiency. “Expected” hormone ranges are seen in Table 1. We offer the following theories:
- ACTH levels were normal despite the presence of enlarged adrenal glands and bilateral TARTs, which are consequences of chronically elevated ACTH levels. The exogenous dexamethasone prescribed to the patient by his local ED likely induced negative feedback on ACTH production by the anterior pituitary gland.
- Renin, aldosterone, and cortisol levels were normal, likely because the patient had the non-classic CAH (NCCAH) that produced some, albeit less efficient, forms of 21-hydroxylase. A sufficient amount of cortisol and aldosterone would therefore be produced, allowing aldosterone to provide negative feedback on renin release. The fact that the patient did not have episodes of adrenal insufficiency despite discontinuing steroid therapy at 18 years old support this theory.
- Normal cortisol levels may also be explained by the presence of a functional ACC as 50 to 80% of hormone-secreting ACCs are cortisol-producing [12]. The results of the LDDST support this theory, as normal cortisol levels indicate a Cushing syndrome where cortisol is produced in excess by some endogenous source [12]. Impaired 21-hydroxylase activity in the setting of NCCAH would counteract the excess production and possibly explain the normal cortisol level.
- Testosterone was low despite its 17-OHP and androstenedione precursors being significantly elevated as expected in 21-hydroxylase deficiency. The downstream deficiency of testosterone production can be attributed to the TARTs that replaced normal testicular tissue with non-functioning heterogeneous material.
Although adrenocortical carcinomas are most often sporadic with an incidence of 0.5 to 7 cases per million per year in the general population, there is evidence for hereditary risk factors [11]. Adrenocortical carcinomas are associated with genetic cancer syndromes such as Beckwith-Wiedemann, Li-Fraumeni, Lynch, Multiple Endocrine Neoplasia-1 (MEN-1), and Familial Adenomatous Polyposis (FAP) [11]. Such hereditary associations raise suspicion when considering the patient’s robust family history of various cancers: the father had colon cancer which may be associated with FAP or Lynch, and the brother had pancreatic cancer which may be associated with MEN-1. Therefore, an unknown number of familial genetic mutations combined with underlying 21-hydroxylase deficiency may be culpable for tumorigenesis and early metastasis seen in our patient.
A genetic component may explain our patient’s accelerated presentation with late signs of multifocal mass effect by 34 years old, which is much younger than the average age of myelolipoma diagnosis at 51 years according to one longitudinal study [13]. TARTs, however, are commonly observed early in life as other studies report the average age of diagnosing TARTs to be 13 years of age without clear correlation to compliance with steroid therapy [8].
In conclusion, CAH is a rare genetic disease with variable presentation. Even rarer is the development of ACC in the setting of underlying CAH, with only a few cases reported. Given our patient’s rapidly progressing malignancy and fatality relatively early in life, we offer two recommendations for patients diagnosed with CAH. First, those with a family history of cancer should be offered genetic screening for mutations associated with, but not limited to, FAP, MEN-1, Beckwith-Wiedmann, Li-Fraumeni, and Lynch to gauge their risk for developing ACC. Second, a positive finding of genetic susceptibility to such syndromes should be used to educate patients on the potential for a poor prognosis if CAH is left untreated and to encourage compliance with long-term steroid therapy to mitigate such risk.
- Burdea, Liliana, Magda DM (2019) "21 Hydroxylase Deficiency - StatPearls - NCBI Bookshelf." Treasure Island (FL): StatPearls Publishing.
- White PC, Speiser PW (2000) Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine reviews 21: 245-291.
- Choi JH, Yoo HW (2017) Management issues of congenital adrenal hyperplasia during the transition from pediatric to adult care. Korean J Pediatr 60: 31-37.
- Yau M, Gujral J, New MI (2019) Congenital adrenal hyperplasia: diagnosis and emergency treatment. InEndotext. MDText. com, Inc.
- Pang S, Becker D, Cotelingam J, Foley TP, Drash AL (1981) Adrenocortical tumor in a patient with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics 68: 242-246.
- Nermoen I, Følling I, Vegge K, Larmo A, Nedrebø BG, et al. (2009) Two adults with adrenal myelolipoma and 21-hydroxylase deficiency. Case Rep Med.
- Chaudhari M, Johnson EK, DaJusta D, Nahata L (2018) Testicular adrenal rest tumor screening and fertility counseling among males with congenital adrenal hyperplasia. J Pediatr Urol 14: 155-e1.
- Kocova M, Janevska V, Anastasovska V (2018) Testicular adrenal rest tumors in boys with 21-hydroxylase deficiency, timely diagnosis and follow-up. Endocr Connect 7: 544-552.
- Gu W, Li L, Dou J, Yang G, Jin N, et al. (2016) Case Report Congenitaladrenal hyperplasia initially presenting with massive adrenal indcidentalomas: a series of 4 cases. Int J Clin Exp Med 9: 13309-13318.
- Taran I, Elder JS (2006) Results of orchiopexy for the undescended testis. World J Urol 24: 231-239.
- Nermoen I, Rørvik J, Holmedal SH, Hykkerud DL, Fougner KJ, et al. (2011) High frequency of adrenal myelolipomas and testicular adrenal rest tumours in adult Norwegian patients with classical congenital adrenal hyperplasia because of 21‐hydroxylase deficiency. Clin Endocrinol 75: 753-759.
- Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, et al. (2014) Adrenocortical carcinoma. Endocr Rev 35: 282-326.
- Bokhari MR, Zulfiqar H, Garla VV (2020) Adrenal Myelolipoma. Treasure Island (FL): StatPearls Publishing.