The inherent diagnostic and therapeutic challenges found in investigation of mitral valve prolapse (MVP) represent a remarkable history within the collective forces of medicine. Beginning with the “new” technology of cardiac ultrasound introduced in the 1950’s, MVP’s clinical relevance has represented an exciting paradigm of how technology identifies but then demands precision in the germane characterization of novel variations from the norm. Abnormalities in the anatomic structure, leaflet function and supportive left ventricular tissue have represented a compilation of sustained efforts to understand clinical outcomes; a quest that continues presently. At one time it was estimated that 5-15% of young women had MVP with some reports suggesting that as many as 35% of patients showed evidence of MVP. Efforts to introduce a more exact definition of MVP, to understand the unique valvular geometry and to link these observations into a coherent appreciation of clinical outcomes has enabled continuing progress and significant reassessment of the numbers of patients affected by MVP. This review briefly summarizes these events and explores the newest areas of investigative effort being undertaken to more effectively assist patients and to avoid the most serious clinical outcomes including complex life-threatening ventricular dysrhythmias and sudden cardiac death.
MVP, myocardium
The history of mitral valve prolapse (MVP) is a fascinating journey through Medicine’s collective application of new technology coupled to an evolving appreciation of fundamental pathophysiology [1]. While this may oversimplify a subject of ever-increasing recognized complexity, it is a reasonable historical summation of events. The advent of the widespread clinical use of cardiac ultrasound in the 1970’s resulted in detection of a presumed novel abnormality involving the mitral valve (MV) predominantly in young women and older patients. At one time it was estimated that 5-15% of young women had MVP with some reports suggesting that as many as 35% of patients showed evidence of MVP [2,3]. It is now felt that these numbers significantly overestimated the prevalence of the disorder and such results were a combination of the exuberant application of new technology, the somewhat ambiguous definitions of what constituted MVP and the erroneous assumption that the MV geometry was flat [4]. This last supposition dictated therefore that any sonographic view that demonstrated excursion of the leaflets of the MV, being superior to the mitral annulus was considered abnormal. Far from a simple planar or two-dimensional construct, the complexity of the mitral annulus was demonstrated by three-dimensional (3-D) echocardiographic imaging and later reconfirmed by cardiac magnetic resonance imaging to be configured in 3-dimensional space as saddle-shaped [5,6].
Refinement in the Definition of “Prolapse”
This detailed work by Levine and others thus resulted in a more accurate understanding of the anatomic proportionality of the in vivo mitral annular shape to its leaflets leading to a more precise definition of MVP [4]. Accordingly, the prevalence of the disorder dropped to what is now felt a more realistic 2-3% of the general population [7]. The actual evolving definition is now recognized to be a single or bi-leaflet prolapse of at least 2 mm beyond the long-axis annular plain, with or without leaflet(s) thickening of greater than 5mm [8]. The presence of leaflet thickening is called classic prolapse and variations of any lesser degrees of leaflet thickening are non classic prolapse [4,9]. Although MVP was in part “popularized” by the advent of widely available transthoracic echocardiography (TTE), it is now recognized that this imaging modality actually may not adequately visualize the entire mitral valve [9,10]. This is at least in part secondary to the afore noted complexity of the inherent geometry of the valve and its supporting apparatus. Charpentier, using the inherent subdivisions or natural boundaries found on each of the MV’s leaflet surfaces has labeled each of these scallops [11]. Both the anterior and posterior leaflets have 3 scallops; lateral (P1), middle (P2) and medial (P3) for the posterior and lateral (A1), middle (P2) and medial (P3) for the anterior [11]. While there are additional nomenclatures identifying scallops based on their respective chordal attachments, these are not as widely used.
Most often the middle posterior leaflet is the prolapsing segment, and this is frequently identified on the long-axis images on TTE. However, identification of the lateral posterior lateral scallop (P1) is not as clearly visualized on this long-axis imaging plane, and verification of its involvement may require use of trans-esophageal echocardiography (TE) [9]. Use of 3-D echocardiography (echo) may further assist as it offers the advantage of simulating the surgeon’s view of the MV but is usually not required to specify the leaflet abnormality. However, 3-D ultrasound imaging is more frequently used to enhance specificity in detection of suspected subtle multi-scallop prolapse. Here a well-defined single scallop prolapse may partially obscure subtle or partial adjoining scallop involvement not readily appreciated by conventional imaging.
The newest imaging modality being employed for MVP is cardiac magnetic resonance (CMR) that allows detailed noninvasive imaging and provides excellent special and temporal resolution [12]. But more importantly, more recent publications support that late gadolinium enhancement (LGE) has been shown in various locations in the left ventricle (LV) and papillary muscle (PM) tips of those with MVP while being absent in controls [12-14]. The enhancement of gadolinium later in the imaging protocol correlates to the presence of fibrotic tissue. This observation has fostered various hypotheses, including speculation that the abnormal motion and force of repetitive traction on MV structures has caused scaring of the valvular apparatus as evidenced by LGE, which is not found in normal volunteers [13,15]. Further, this observation has been linked to potential determinative clinical outcomes of lowered dysrhythmia thresholds. This latter concern is of great interest to investigators as the previously benign reputation of MVP, still prevalent within healthcare, has been supplanted with recognition of the increased risk of ventricular dysrhythmias and sudden cardiac death (SCD) in specific subsets of patients with authenticated MVP [13,16].
More recent recognition that MVP is associated with SCD in 7% of young adults, 13% being young women with the latter representing those at particular risk of ventricular arrhythmias [13]. This higher risk group also being found clinically to have a mid-systolic click on auscultation, bi-leaflet involvement, T-wave abnormalities on inferior leads and right bundle branch block (RBBB)-type or polymorphic ventricular arrhythmias on electrocardiograms (ECGs) [13]. But it is the previously mentioned LGE on CMR that correlated to myocardial fibrosis, confirmed histologically in SCD victims, which underlined a reason for inherent electrical instability and dysrhythmia proclivity [13,15,16]. This finding underwrites the often unrecognized and certainly under-appreciated linkage of MVP to SCD. Prevalence rates for MVP being linked to SCD vary widely, ranging from 0-24%, the expanded range likely resulting from the inconsistent diagnostic criteria for MVP noted above [13,17]. In addition, the benign historical reputation of MVP is believed to have resulted in SCD potentially attributable to MVP not being aggressively examined pathologically, ultimately overlooking the macroscopic and histological evidence of significant scaring and dysrhythmia potential [18]. Importantly, the association between MVP and SCD is not only limited to those patients that have significant mitral insufficiency, as hemodynamic valvular regurgitation reportedly increases the occurrence of ventricular dysrhythmias. But more recent data has noted that PVCs may originate in the mitral annulus, the LV outflow tract and the PMs and thus these structures close to the prolapsing leaflets are themselves putative dysrhythmic triggers [19]. But this remains a point of speculation joining a list of other MVP-related ventricular dysrhythmia triggering factors, including but not limited to excessive traction on the PMs, elongated chordae with afterdepolarization-induced triggered activity and even diastolic depolarization of muscle fibers in redundant leaflets with triggered repetitive automaticity [1,20,21] (Figure 1).
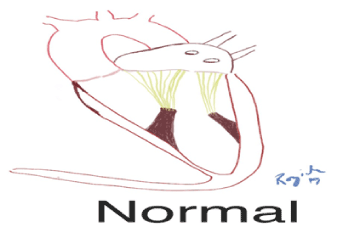
Figure 1 . Schematic of normal mitral valve with leaflets coaptation point at the annulus of the mitral valve apparatus.
MVP have Sudden Cardiac Death? Is it the myocardium?
Examining the LV myocardium as a potential source of dysrhythmias has been a relatively new occurrence in those suffering SCD in the setting of existing MVP. While previous pathological reports identified valvular structural abnormalities, including alterations in annular circumference, leaflet length and thickness along with the presence and extent of endocardial plaques, little attention was focused upon the LV myocardium itself [22-26]. The LV myocardial scarring observed however, is qualitatively different from that observed in ischemic heart disease, as this latter pattern of fibrosis is regionalized to the specific area of ischemic injury and is relatively uniform in the histological appearance [27]. This contrasts with newer reports that detailed the fibrotic characteristics of MVP-SCD victims, in that affected areas were “patchy” with both fibrotic and healthy myocardial cells co-existing within identical regions of sampled tissue [14,28]. These studies while attempting to add some clarity to LV myocardial involvement, as they specified that there was actual scarring in PMs and the posterolateral wall, have conflicted with other very recent reports supporting a potential MVP-cardiomyopathy [13,14]. Important also, is the distinction that CMR-LGE identification of abnormal LV myocardium as a source for MVP-SCD, is independent of moderate to severe mitral regurgitation [19]. This lack of valvular regurgitant correlation alters the previous somewhat narrow scope of understanding that focused on the principal role that inherent MV-regurgitant pathology, linked to significant hemodynamic effects, were causally related to SCD in the setting of MVP. It now appears that intrinsic alteration of the LV myocardial cellular structure itself, with LGE-detectable scarring, contributes to the increase in SCD associated in otherwise healthy individuals with MVP [13,14,29].
This distinction is relevant as it may realign past explanations of SCD associated with MVP and initiate requisite changes in diagnostic and therapeutic responses to improve outcomes. First, the early descriptions of abnormal LV contraction patterns coupled to ECG abnormalities hypothesized that MVP was indeed constituted by an abnormal myocardium [30,31]. Different labels emerged but the so-called” MVP Syndrome” advanced the concept that a cardiomyopathy with heterogeneous regions of myocardial contractility underwrote the ultimate disruption in the mitral valvular apparatus leading to the development of myxomatous tissue and leaflet thickening [30,31]. The suggestion was that this MVP-phenotype involved development of myxomatous tissue and leaflet thickening associated with an inherent cardiomyopathy [14]. Alternative data supported by CMR-LGE argue that the LV myocardial structural pathology, the scarring and fibrotic changes, are a consequence of the MVP itself [13]. In this view the repetitive systolic mechanical stretch of the myocardium directly supporting or interconnected to the valve (PMs and inferobasal wall segments) ultimately hypertrophy beyond sustainable physiologic tolerances resulting in fibrosis. Proponents of this theory cite the increased cardiomyocyte diameter together with site-specific replacement-type fibrosis as supporting evidence [13] (Figure 2).
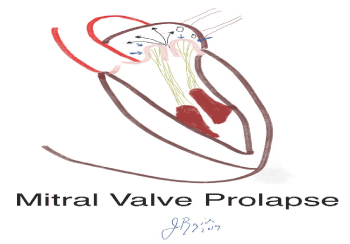
Figure 2 . Schematic of mitral valve prolapse with leaflets dramatically billowing (4 blue arrows) into the left atrium. There is associated significant mitral regurgitation.
Who is Really at Risk?
The suggestion that patients with MVP are indeed at increased risk for SCD, begs the question as to the needed refinements in our examination of subgroups to determine those most vulnerable. High risk groups such as those encountering and surviving SCD clearly require further study and likely AICD implant [32]. But in those with complex ventricular dysrhythmias with unexplained syncope or pre-syncope or including those with ECG depolarization abnormalities in the inferolateral leads or with bi-leaflet prolapse, further testing is also appropriate [16]. These cohorts will likely be suitable for evaluation with CMR and LGE to identify increased LV fibrosis before randomized trials can more definitively explicate which groups conclusively benefit from AICD implant [13]. However, a cautionary note is requisite before concluding that CMR-LGE detection of LV fibrosis can definitively identify those at SCD risk, or that subsequent intervention for LV fibrosis will prevent SCD related to MVP.
The above mentioned repetitive systolic mechanical stretch of structures or regions directly supporting the MV (inferobasal wall segments and PMs) are proposed as sites developing vulnerable fibrotic myocardium [12,13]. It is suggested that the triggered hypertrophy in these regions continues beyond physiologic tolerances causing local injury to functioning myocytes, which are then supplanted by fibrotic dysrhythmia-susceptible tissue. In effect there is scar formation. But triggers for myocardial growth are a complex and nuanced interplay impacting both cellular and molecular elements. Proposed factors, such as physical “load”, or deformity of intact cellular length, surface area and sarcomere length have been shown sufficient to elicit induction of increased RNA and protein synthetic activity in adult mammalian cardiac cells [33]. The interconnecting biological steps can be remarkably subtle with hidden redundancy to ensure normative morphology and function. Disruption of these pathways results in excessive myocardial hypertrophy that is maladaptive to both mechanical and electrical performance. As such, load may be determinative in recruiting early molecular signaling to induce a broader genomic activation leading to myocyte and interstitial hypertrophic growth [34]. But whether this leads to discrete “scar” formation that is determinative in SCD associated with MVP is at present entirely speculative. Alternatively, myocardial hypertrophic growth may selectively alter the normal ratios of numerical populations for myocyte-interstitial cells, as the added interstitial cell infrastructure forms the lattice of support for the larger myocytes exposed to pressure overloading [14,35-37]. The actual numeric variance from normal for the increased interstitial cell populations may lead to disruption of electrochemical conduction causing the recognized propensity to dysrhythmias. The collective change in interstitial mass associated with pressure-loaded hypertrophic growth is well described [14,35-37]. Thus, direct physical loading or pressure changes impacting myocardial structures may act as a “trip wire” initiating cellular and molecular activation that if sustained will geometrically alter (hypertrophy) the myocardium [35,38-40]. Each of these cellular variances may then recruit or magnify paracrine, autocrine and endocrine influences that further bend from normal the eventual physical appearance and physiologic outcome [41]. As noted, some argue that this ultimately results in the recognizable hypertrophy followed by proposed fibrotic scarring (PM or inferolateral fibrosis) [42]. This fibrotic change and scaring have thus been inferred as creating the inherent vulnerability to electrical instability. But it may be important to carefully examine the linkage of this “causality” between the identified hypertrophic LV remodeling and the hypothesized source of dysrhythmic potential.
What Causes LV Fibrotic Change and Is It Important?
Fibrotic changes and scar are detected by CMR-LGE with the understanding that larger areas of LGE change predict poorer long-term prognosis. However, MVP associated dysrhythmias may actually require neither fibrosis or scarring. Reports have shown that MVP-SCD risk is also a consequence of severe mitral valvular hemodynamic pathology independently generating malignant dysrhythmias [43]. This offers a different mechanism of arrhythmogenic etiology than LV fibrosis and scar detectable by LGE. Further, Basso et al suggested that the complex effect of mechanical stretch by the prolapsing leaflets of the MV and their associated elongated chordae may also act as a trigger for dysrhythmogenic pathology. Thus, two potentially disparate but important mechanisms for dysrhythmias may exist beyond the hypothesis that fibrotic LV is mechanistically critical. It must be asked whether the contribution of LV fibrosis, hemodynamic valvular severity or elongated chordal elements are collectively important, or individually determinative as the principal triggers in SCD associated with MVP? Reliance on LGE to identify fibrotic areas may indeed reliably detect specific cohorts at increased risk for MVP-associated SCD [14]. But as noted, SCD related to LV fibrosis and scar in the setting of MVP may be but one of several arrhythmogenic mechanisms. Thus LGE-identified cohorts may inadequately quantify the totality of those at risk.
But even those who have MVP-associated LV remodeling and subsequent fibrosis, it is presently unknown what amount of fibrosis or even what histological pattern or composition of fibrotic change paralleling SCD susceptibility is going to be detectable by LGE [14]. Undoubtedly, there are limits to the LGE identification of fibrosis as sensitivity and specificity for CMR-LGE fibrotic detection is still evolving [44-46]. Alternatively, it may be that an “MVP phenotype” with increased myxomatous tissue and leaflet thickening mentioned previously predisposes to regional heterogeneity of LV fibrotic tissue absent any discrete LV scarring mechanistically tied to MVP stress or injury [9]. It may even be that the myxomatous tissue may inherently “mature” to have aberrant numerical ratios of myocyte-interstitial cell populations and may hint at why fibrosis in some patients with MVP is diffuse and ill-defined [14,47]. This last conceptual entity, that of progression of tissue change over time has gained potential traction as Delling et al. have noted that nondiagnostic MVP morphologies (NDM) may “evolve” to recognizable phenotypic MVP or 3-16 years in affected pedigrees [9]. Thus, hypothetical constructs of mechanistically important “wear and tear” related to valvular motion and stress causing discrete scar versus an MVP phenotype “evolving” in predisposed genotypic clusters may compete for relevance or represent but two of a group of multiple MVP clinical presentations. Thus, substantive MVP phenotypic variance includes those with myocardial cells undergoing hypertrophic growth prior to fibrotic replacement and discrete scar formation in addition to an MVP-cardiomyopathy [14]. Whether fibrotic scar detectable by LGE is the predominant marker for MVP-SCD remains unknown. These are relevant concerns as each possibility may impact the spatial and temporal resolution for fibrotic detection in CMR-LGE and may dictate one of several strategies for therapeutic intervention.
Clinical Relevance
What all of this potentially means for the practicing clinician, who in 2019 is caring for a patient labeled as having MVP, is instructive. First, based on the timeframe that the patient was first diagnosed with MVP, clinicians have to confirm that the diagnosis is correct. For patients being diagnosed in the 1970s to the early 2000’s, the residual effects of laxity in the pathological definition often clouds the requisite precision provided by Levine [4]. Repeating a detailed echocardiogram is essential and failure to meet modern definitions of MVP are not uncommon. Second, and this now begins the more challenging task for clinicians, is once the classic or non-classic MVP is identified as defined by modern criteria, who within this group is at risk of a variety of adverse sequelae? The practical initial bifurcating point is whether significant mitral regurgitation coexists with the MVP. This may independently require surgical intervention as supported by ACC guidelines for MR/MVP [8]. But in patients with no significant hemodynamic abnormalities of the mitral valve, a comprehensive assessment of past history, such as unexplained syncope or symptomatic palpitations is required. But further detailed and individualized care may call for dysrhythmia monitoring or CMR looking for subtle structural or fibrotic changes. It is at this point that a team approach offers the greatest value as electrophysiologic, surgical and imaging expertise may optimize outcomes. The “story” of MVP is a unique tale within the confines of Medicine, it is far from complete.
- Barlow JB, Bosman CK (1966) Aneurysmal protrusion of the posterior leaflet of the mitral valve. An auscultatory-electrocardiographic syndrome. Am Heart J 71: 166-78.
- Savage DD, Devereux RB, Garrison RJ, Castelli WP, Anderson SJ, et al. (1983) Mitral valve prolapse in the general population. 2. Clinical features: the Framingham Study. Am Heart J 106: 577-581. [Crossref]
- Savage DD, Garrison RJ, Devereux RB, Castelli WP, Anderson SJ, et al. (1983) Mitral valve prolapse in the general population. 1. Epidemiologic features: the Framingham Study. Am Heart J 106: 571-576. [Crossref]
- Levine RA, Stathogiannis E, Newell JB, Harrigan P, Weyman AE (1988) Reconsideration of echocardiographic standards for mitral valve prolapse: lack of association between leaflet displacement isolated to the apical four chamber view and independent echocardiographic evidence of abnormality. J Am Coll Cardiol 11: 1010-1019. [Crossref]
- Naoum C, Blanke P, Cavalcante JL, Leipsic J (2017) cardiac computed tomography and magnetic resonance imaging in the evaluation of mitral and tricuspid valve disease: Implications for transcatheter interventions. Circ Cardiovasc Imaging 10: 1-10.
- Thavendiranathan P, Phelan D, Thomas JD, Flamm SD, Marwick TH (2012) Quantitative assessment of mitral regurgitation: validation of new methods. J Am Coll Cardiol 60: 1470-1483.
- Delling FN, Rong J, Larson MG, Lehman B, Fuller D, et al. (2016) Evolution of mitral valve prolapse: insights from the framingham heart study. Circulation 133: 1688-1695. [Crossref]
- American College of C, American Heart Association Task Force on Practice G, Society of Cardiovascular A (2006) ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing Committee to Revise the 1998 guidelines for the management of patients with valvular heart disease) developed in collaboration with the Society of Cardiovascular Anesthesiologists endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J Am Coll Cardiol 48: e1-148.
- Delling FN, Vasan RS (2014) Epidemiology and pathophysiology of mitral valve prolapse: new insights into disease progression, genetics, and molecular basis. Circulation 129: 2158-2170.
- Foster GP, Isselbacher EM, Rose GA, Torchiana DF, Akins CW, et al. (1998) Accurate localization of mitral regurgitant defects using multiplane transesophageal echocardiography. Ann Thorac Surg 65: 1025-1031. [Crossref]
- Carpentier A (1983) Cardiac valve surgery--the "French correction". J Thorac Cardiovasc Surg 86: 323-337. [Crossref]
- Han Y, Peters DC, Salton CJ (2008) Cardiovascular magnetic resonance characterization of mitral valve prolapse. JACC Cardiovasc Imaging 1: 294-303.
- Basso C, Perazzolo Marra M, Rizzo S (2015) Arrhythmic mitral valve prolapse and sudden cardiac death. Circulation 132: 556-66.
- Garbi M, Lancellotti P, Sheppard MN (2018) Mitral valve and left ventricular features in malignant mitral valve prolapse. Open Heart 5: e000925. [Crossref]
- Sriram CS, Syed FF, Ferguson ME (2013) Malignant bileaflet mitral valve prolapse syndrome in patients with otherwise idiopathic out-of-hospital cardiac arrest. J Am Coll Cardiol 62: 222-230.
- Han HC, Ha FJ, Teh AW (2018) Mitral valve prolapse and sudden cardiac death: a systematic review. J Am Heart Assoc 7: e010584.
- Basso C, Calabrese F, Corrado D, Thiene G (2001) Postmortem diagnosis in sudden cardiac death victims: macroscopic, microscopic and molecular findings. Cardiovasc Res 50: 290-300.
- Basso C, Burke M, Fornes P, Gallagher PJ, de Gouveia RH, et al. (2008) Guidelines for autopsy investigation of sudden cardiac death. Virchows Arch 452: 11-18. [Crossref]
- Kligfield P, Levy D, Devereux RB, Savage DD (1987) Arrhythmias and sudden death in mitral valve prolapse. Am Heart J 113: 1298-1307. [Crossref]
- Cobbs BW, King SB (1977) Ventricular buckling: a factor in the abnormal ventriculogram and peculiar hemodynamics associated with mitral valve prolapse. Am Heart J 93: 741-58.
- Salazar AE, Edwards JE (1970) Friction lesions of ventricular endocardium. Relation to chordae tendineae of mitral valve. Arch Pathol 90: 364-376.
- Chesler E, King RA, Edwards JE (1983) The myxomatous mitral valve and sudden death. Circulation 67: 632-639. [Crossref]
- Davies MJ, Moore BP, Braimbridge MV (1978) The floppy mitral valve. Study of incidence, pathology, and complications in surgical, necropsy, and forensic material. Br Heart J 40: 468-481. [Crossref]
- Dollar AL, Roberts WC (1991) Morphologic comparison of patients with mitral valve prolapse who died suddenly with patients who died from severe valvular dysfunction or other conditions. J Am Coll Cardiol 17: 921-931. [Crossref]
- Farb A, Tang AL, Atkinson JB, McCarthy WF, Virmani R (1992) Comparison of cardiac findings in patients with mitral valve prolapse who die suddenly to those who have congestive heart failure from mitral regurgitation and to those with fatal noncardiac conditions. Am J Cardiol 70: 234-239.
- Pocock WA, Bosman CK, Chesler E, Barlow JB, Edwards JE (1984) Sudden death in primary mitral valve prolapse. Am Heart J 107: 378-382. [Crossref]
- Friedrich MG (2008) Tissue characterization of acute myocardial infarction and myocarditis by cardiac magnetic resonance. JACC Cardiovasc Imaging 1: 652-662.
- John BT, Tamarappoo BK, Titus JL, Edwards WD, Shen WK, et al. (2004) Global remodeling of the ventricular interstitium in idiopathic myocardial fibrosis and sudden cardiac death. Heart Rhythm 1: 141-149.
- Lichstein E (1980) Site of origin of ventricular premature beats in patients with mitral valve prolapse. Am Heart J 100: 450-457. [Crossref]
- Crawford MH, O'Rourke RA (1984) Mitral valve prolapse: a cardiomyopathic state? Prog Cardiovasc Dis 27: 133-139. [Crossref]
- Gulotta SJ, Gulco L, Padmanabhan V, Miller S (1974) The syndrome of systolic click, murmur, and mitral valve prolapse--a cardiomyopathy? Circulation 49: 717-728. [Crossref]
- Vaidya VR, DeSimone CV, Damle N, Naksuk N, Syed FF, et al. (2016) Reduction in malignant ventricular arrhythmia and appropriate shocks following surgical correction of bileaflet mitral valve prolapse. J Interv Card Electrophysiol 46: 137-143. [Crossref]
- Mann DL, Kent RL, Cooper Gt (1989) Load regulation of the properties of adult feline cardiocytes: growth induction by cellular deformation. Circ Res 64: 1079-1090.
- Cooper Gt (2000) Cardiocyte cytoskeleton in hypertrophied myocardium. Heart Fail Rev 5: 187-201.
- Cooper G, Kent RL, Mann DL (1989) Load induction of cardiac hypertrophy. J Mol Cell Cardiol 21: 11-30. [Crossref]
- Cooper Gt, Mercer WE, Hoober JK (1986) Load regulation of the properties of adult feline cardiocytes. The role of substrate adhesion. Circ Res 58: 692-705.
- Cooper G, Tomanek RJ (1982) Load regulation of the structure, composition, and function of mammalian myocardium. Circ Res 50: 788-798. [Crossref]
- Jones LG, Rozich JD, Tsutsui H, Cooper Gt (1992) Endothelin stimulates multiple responses in isolated adult ventricular cardiac myocytes. Am J Physiol 263: 1447-1454.
- Kent RL, Rozich JD, McCollam PL, McDermott DE, Thacker UF, et al. (1993) Rapid expression of the Na(+)-Ca2+ exchanger in response to cardiac pressure overload. Am J Physiol 265: 1024-1029. [Crossref]
- Rozich JD, Barnes MA, Schmid PG, Zile MR, McDermott PJ, et al. (1995) Load effects on gene expression during cardiac hypertrophy. J Mol Cell Cardiol 27: 485-99.
- Carreño JE, Apablaza F, Ocaranza MP, Jalil JE (2006) Cardiac hypertrophy: molecular and cellular events. Rev Esp Cardiol 59: 473-486. [Crossref]
- Tagawa H, Rozich JD, Tsutsui H, Narishige T, Kuppuswamy D, et al. (1996) Basis for increased microtubules in pressure-hypertrophied cardiocytes. Circulation 93: 1230-1243. [Crossref]
- Turker Y, Ozaydin M, Acar G (2010) Predictors of ventricular arrhythmias in patients with mitral valve prolapse. Int J Cardiovasc Imaging 26: 139-145.
- Kadish AH, Bello D, Finn JP (2009) Rationale and design for the Defibrillators to Reduce Risk by Magnetic Resonance Imaging Evaluation (DETERMINE) trial. J Cardiovasc Electrophysiol 20: 982-987.
- Schmidt A, Azevedo CF, Cheng A, Gupta SN, Bluemke DA, et al. (2007) Infarct tissue heterogeneity by magnetic resonance imaging identifies enhanced cardiac arrhythmia susceptibility in patients with left ventricular dysfunction. Circulation 115: 2006-2014. [Crossref]
- Disertori M, Rigoni M, Pace N (2016) Myocardial fibrosis assessment by lge is a powerful predictor of ventricular tachyarrhythmias in ischemic and nonischemic lv dysfunction: A meta-analysis. JACC Cardiovasc Imaging 9: 1046-1055.
- Bui AH, Roujol S, Foppa M (2017) Diffuse myocardial fibrosis in patients with mitral valve prolapse and ventricular arrhythmia. Heart 103: 204-209.