Background: Chronic myeloproliferative neoplasms (MPN) are clonal disorders characterized by an increased risk of thrombosis and of leukemic evolution. Paroxysmal nocturnal hemoglobinuria (PNH) is a rare disease whose characteristic traits are intravascular hemolysis and thrombophilia. The PNH clone is classically associated with bone marrow failure syndromes and, accordingly, it is thought to expand because the remaining hematopoiesis is suppressed. Conversely, here we report the detection of a PNH population in 2 MPN patients presenting with thrombosis.
Case presentation: The first case is a 72-year-old woman presenting with a portal vein thrombosis associated with leukothrombocytosis and anemia; additional investigations diagnosed a JAK2-mutated essential thrombocythemia in myelofibrotic evolution and a significant PNH population. The second case is a 75-year-old man with JAK2-mutated polycythemia vera and a long history of thrombosis, who was found to carry a small PNH clone. Both our patients have experienced severe thrombotic events, while neither of them has ever reported signs of overt hemolysis.
Conclusions: These cases defy the most accepted etiopathogenetic theory for PNH, challenge the classical indications for PNH screening and raise several practical questions, such as the clinical significance of small PNH clones and the management of patients with two pro-thrombotic factors with different etiologies.
myeloproliferative neoplasms, paroxysmal nocturnal hemoglobinuria, thrombosis
Chronic myeloproliferative neoplasms (MPN) are a group of clonal hematopoietic disorders that include polycythemia vera (PV), essential thrombocythemia (ET) and myelofibrosis (MF). They are all clinically characterized by the proliferation of one or more myeloid lineages, and are associated with an increased risk of thrombosis and of leukemic evolution.
Paroxysmal nocturnal hemoglobinuria (PNH) is another clonal hematopoietic disorder, caused by the somatic mutation of the PIG-A gene, leading to the absence of GPI-anchored proteins at the cell surface. These features make red blood cells more prone to be attacked by the complement system, resulting in intravascular hemolysis, which is the hallmark of PNH; moreover, free hemoglobin, PNH platelets activation by the complement system and other mechanisms less well understood are responsible for thrombophilia, the other characteristic trait of PNH [1].
Even though PNH is a clonal disorder, it is classically considered benign or non-neoplastic; in fact, the PNH clone is thought to expand because the remaining hematopoiesis is suppressed by the immune system, that seems to spare PNH cells. In other words, the PNH clone would have an extrinsic growth advantage. This phenomenon could explain the strong correlation between PNH and bone marrow failure syndromes, especially aplastic anemia (AA) and myelodysplastic syndromes (MDS) [2].
However, PNH clones are occasionally found in non-cytopenic patients, either in patients with no evident bone marrow failure syndrome, either after a complete recovery from AA. Moreover, recent case reports described sporadic patients presenting with MPN and PNH with overt hemolysis [3,4]. At the same time, from the molecular point of view, Shen et al. [5] found that several somatic mutations can arise in the PNH clone, possibly resulting in an intrinsic growth advantage.
Here we report the unexpected detection of a PNH population in 2 MPN patients presenting with thrombosis but without overt hemolysis.
Case #1
The first case is a 72-year-old woman presenting to the emergency department for severe abdominal pain. Her past medical history was insignificant. An abdominal computed tomography (CT) scan with intravenous contrast showed a portal vein thrombosis and a large splenic lesion possibly compatible with a splenic infarct (Figure 1).
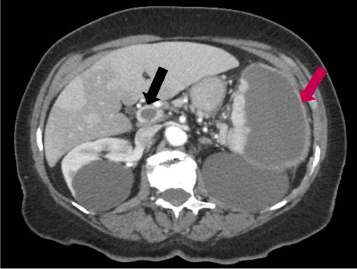
Figure 1. Abdominal CT scan of patient # 1. The black arrow indicates the portal thrombus; the purple arrow indicates the splenic lesion.
Her blood exams revealed neutrophilic leukocytosis (WBC 29.350 x 109/L, neutrophils 83.3%), thrombocytosis (PLT 827 x 109/L) and anemia (Hb 7.6 g/dL); LDH was markedly elevated (656 U/L), INR was 1.54, while aPTT ratio was 1.07; haptoglobin was consumed (< 0.08 g/L), but reticulocytes and bilirubin were normal, as well as her urine analysis, B12 and folic acid levels.
She was hospitalized in our Hematological Division and initiated to low molecular weight heparin. The bone marrow biopsy was diagnostic for an ET in myelofibrotic evolution; accordingly, a JAK2 V617F mutation was found in 19.88% of alleles on peripheral blood. Moreover, the flow cytometry analysis performed on peripheral blood also showed a significant PNH population: 88.6% of granulocytes, 86.9% of monocytes and 71% of red blood cells were FLAER negative (i.e., PNH cells). Surprisingly, her urine analysis was completely normal, she had never experienced dark urine episodes nor any other classical PNH sign or symptom.
The patient was treated with hydroxyurea and low molecular weight heparin, then switched to warfarin; progressively, the count of leukocytes and platelets, as well as LDH and hemoglobin levels, almost normalized; moreover, the number of reticulocytes and the bilirubin level continued to be normal, while haptoglobin was still consumed (Table 1). The portal vein was recanalized within 2 months.
Table 1. Timeline case #1. WBC: white blood cells count; N: neutrophils; Hb: hemoglobin; PLTs: platelets; LDH: lactate dehydrogenase; HU: hydroxyurea; LMWH: low molecular weight heparin. Normal ranges are written in square brackets.
Case # 1 |
WBC (N%) [4.4-11 x 109/L] |
Hb [12-16 g/dL] |
PLT [150-400 x 109/L] |
LDH [150-300 U/L] |
Haptoglobin [0, 5-2 g/L] |
Urine analysis |
PNH population |
At diagnosis |
29.350 x 109/L (N 83,3%) |
7.6 g/dL |
827 x 109/L |
656 U/L |
< 0.08 g/L |
Normal (no Hb) |
88.6% of granulocytes, 86.9 % of monocytes, 71% of red blood cells. |
2 weeks after (HU and LMWH ongoing) |
12.3 x 109/L (N 66,7%) |
9.2 g/dL |
758 x 109/L |
332 U/L |
< 0.08 g/L |
Normal (no Hb) |
Not evaluated. |
9 months after (HU and warfarin ongoing) |
7.13 x 109/L (N 51,6%) |
12.6 g/dL |
424 x 109/L |
324 U/L |
< 0.08 g/L |
Normal (no Hb) |
80 % of granulocytes, 75.5% of monocytes, 45% of red blood cells. |
Nine months after diagnosis, we performed a complete revaluation of the patient (Table 1). We found that the PNH clone was substantially stable, affecting 80% of granulocytes, 75.5% of monocytes and 45% of red blood cells were; the JAK2 mutation was found in 7.5% of the alleles in peripheral blood leukocytes (Table 1).
Subsequently, we performed a flow cytometric sorting of the patient’s peripheral blood cells, isolating 4 different populations (PNH positive and negative granulocytes, PNH positive and negative monocytes) and then tested for the JAK2 mutation. We discovered that the PNH positive populations, both granulocytes and monocytes, were enriched in JAK2 mutated cells (13.02% and 9.37% of the alleles respectively), while both PNH negative populations showed a low positivity for the JAK2 mutation, which correlated with the contamination occurred during the sorting. These results suggest that the JAK2 positive subclone had arose within the PNH population.
Case #2
The second case is a 75-year-old man, with a long history of both arterial and venous thrombosis, as shown in Figure 2.
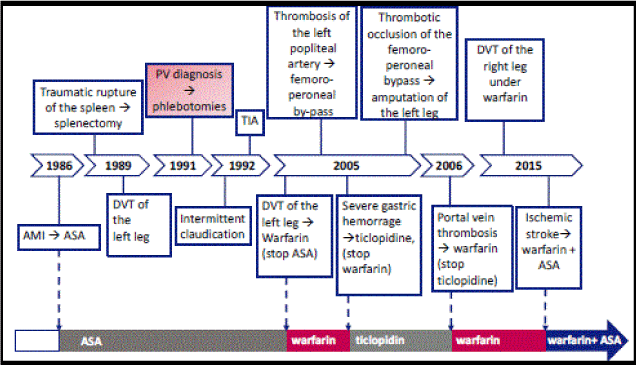
Figure 2. Timeline of thrombotic and bleeding events of patient # 2. AMI: acute myocardial infarction; ASA: acetylsalicylic acid (low doses); DVT: deep venous thrombosis; PV: polycythemia vera; TIA: transient ischemic attack.
The patient finally arrived at our emergency department on June 2015 for an ischemic stroke. His blood analysis revealed: WBC 8.590 x 109/L, Hb 13.5 g/dL, hematocrit 43%, PLT 429 x 109/L, LDH 409 U/L; INR was 2.04; he was assuming warfarin, but not an antiplatelet drug.
The classical screening for acquired or congenital thrombophilia was negative. Morever, during the two last events, the hematocrit was < 45%, and INR was between 2 and 2.1. We completed the screening looking for a PNH clone, and we actually found a very small PNH population, accounting for 0.05% of granulocytes and 0.13% of monocytes. We analyzed his exams more carefully: his urine analysis was negative for hemoglobinuria; haptoglobin, bilirubin and reticulocytes were normal; the pat ient had never experienced dark urine episodes.
We monitored this small clone at 6 and 18 months, and it was persistent and quantitatively stable; no laboratory or clinical sign of hemolysis was ever found.
The patient himself had noted that all his thrombotic events had occurred when INR was around 2; we decided, together with the cardiologist, the neurologist and the patient, to try to maintain INR between 2.5 and 3.5, and to start again an antiplatelet drug; he is still assuming hydroxyurea, and his blood count is perfectly controlled. He has not experienced any thrombotic or bleeding event in the last 1,5 years.
Considering the very small amount of PNH cells, we didn’t try to sort them and look for JAK2 V617F mutation; however, we repeated the real time quantitative PCR assay for JAK2 mutation, finding that 36.1% of the alleles in peripheral blood leukocytes were positive.
We report here the cases of 2 patients with MPN and a PNH population; both of them have had recurrent or severe/atypical thrombosis, while neither of them has ever reported hemoglobinuria episodes, nor other signs of florid hemolysis or classical PNH features other than thrombosis, such as abdominal pain, dyspnea or chronic kidney disease.
Our two cases, together with others recently reported in the literature [3,4], turn on the spotlights on several open issues:
- When to screen for PNH, i.e., the so called ‘reasons for testing’. Consensus Recommendations [6,7] indicate that patients requiring a PNH screening include those presenting with thrombosis associated with signs of hemolytic anemia or with unexplained cytopenia, or thrombosis at unusual sites (such as splanchnic veins or cerebral sinuses); the latter recommendation has been summarized under the term ‘unexplained thrombosis’ in the Consensus Recommendations text itself [6], and in the subsequent scientific papers or specialized educational material [8-10].
MPN are well known to be associated with unusual thrombosis: for example, approximately 40% of patients with splanchnic vein thrombosis are JAK2 mutated [11]. Following the usual interpretation of the Consensus Recommendations, our patients would have not been screened for PNH: first of all, they presented an explainable thrombosis; moreover, the first case just had anemia, which is compatible with myelofibrosis, and the second case did not have cytopenias. Furthermore, laboratory tests that can detect a subclinical hemolysis are often found to be altered in MPN: LDH, in particular, is frequently elevated in myelofibrosis, but also in PV and ET; reticulocytes, bilirubin and, to a lesser extent, haptoglobin can be altered in MPN due to a number of factors, such as increased erythroid proliferation or ineffective erythropoiesis, therapy with erythropoietin or hydroxyurea and, in particular cases, splenectomy [12-15].
Altogether, it would be easy for a clinician to ascribe the whole clinical picture to the MPN alone, missing the PNH diagnosis. However, Primignani et al., found that 38.2% of JAK2 mutated patients with a splanchnic vein thrombosis actually presented at least one additional risk factor for thrombosis, such as a hypercoagulable disorder or a predisposing condition [11].
This and other case reports [3,4] show that the association between PNH and MPN is a reality; moreover, we show that MPN can be associated to a PNH clone even if there is no overt hemolysis. However, before any recommendation could be formulated, further investigations are needed to assess the frequency of this association.
- Which clinical implications can have a double diagnosis (i.e., MPN and PNH)? Does the size of the clone matter? And then: which is the clinical significance of a small PNH clone in an MPN patient? Can we still hypothesize a pro-thrombotic synergism? There are currently no answers to these questions.
Both MPN and PNH are ‘thrombophilic diseases’, with the risk to develop a major thrombosis as high as about 50% for both conditions [1,16-19]. Thrombosis in PNH is probably due to a number of mechanisms, such as complement-mediated activation of platelets, Nitrogen Oxide depletion, toxicity of free hemoglobin and increase in inflammatory citokines; interestingly, many of these mechanisms are hemolysis-independent [1], suggesting that even patients without overt hemolysis (such as both our patients) and/or small clones (such as our second patient), could have an increased risk.
However, data analyzing the impact of the size of the clone on the risk of thrombosis are controversial: Hall, et al.[20] found that patients with a larger clone had a greater risk than those with a smaller one; a retrospective analysis of the Korean registry [21], on the contrary, found that there was no relationship between the clone size and the development of thrombotic events. Moreover, the limit between small and large clones is arbitrary; Hall et al, for example, established a 50% threshold [20], while Lee et al. divided the patients in 3 categories (< 20%, 20-50% and > 50%) [21]; no study, at the best of our knowledge, has ever investigated if very small clones (e.g. < 1%, such as our second patient) are responsible for an increased thrombotic risk.
Even more interestingly, anticoagulants reduce the risk of thrombosis in PNH patients [20], but numerous thrombotic events have been described while patients were therapeutically anticoagulated [22,23]. Eculizumab, a complement inhibitor, showed to be effective in reducing the thrombotic rate in PNH patients, even when they were already anticoagulated [23]. However, eculizumab efficacy and safety has never been extensively assessed in MPN patients. Sugimori, et al.reported on 3 cases of JAK2 mutated patients who developed a florid PNH and were treated with eculizumab; one of them developed a Budd Chiari Syndrome under eculizumab and anticoagulant treatment [3], but it has to be said that eculizumab trough levels were low.
2021 Copyright OAT. All rights reserv
A recent meta-analysis suggested that ruxolitinib, a JAK2 inhibitor, could reduce the incidence of thrombosis in PV and MF [24]. However, these results have to be confirmed in prospective trials and, to date, ruxolitinib is not indicated for the sole prevention of thrombosis in MPN patients with high thrombotic risk, as our patients could be considered; obviously, there are no data on the use of ruxolitinib in patients with a PNH population.
In summary, at present, we are not able to quantify the relative additional risk induced by the PNH clone in an MPN patient, especially when the size of the clone is very small; moreover, until we won’t be able to predict the effect of eculizumab and/or ruxolitinib on the thrombotic risk of these patients, no recommendation can be formulated.
- The biological basis of the PNH clone expansion. As already mentioned, the main hypothesis is that the PNH population growth is due to an immune mediated suppression of the GPI positive hematopoiesis, where the GPI anchor would be the autoimmune target for T lymphocytes, probably in an HLA-restricted manner or in a HLA-independent manner [25-27]. However, this extrinsic growth advantage does not occur in MPN where, on the contrary, the bone marrow is generally hypercellular; in these situations, it is more plausible to hypothesize that the PNH clone has an intrinsic growth advantage.
Even more interestingly, the two reported cases show how the PNH and the MPN clone can have different ‘relationships’. In the first case, the JAK2 positive subclone arose within a pre-existing PNH population. In the second case, we couldn’t sort the PNH cells due to the very small size of this clone; however, since 36.1% of the alleles in peripheral blood leukocytes were JAK2 mutated, it is possible to hypothesize 2 scenarios: i) the PNH clone arose in the JAK2 mutated population; ii) the PNH clone arose in parallel to the JAK2 mutated population. The fact that the JAK2 mutation is not always the founding mutation of an MPN, actually complicates the picture.
Nevertheless, our data are consistent with what Shen, et al. [5] found analyzing the clonal architecture of PNH: they showed that the PIG-A mutation could occur alone, or it could be the initial event followed by one or more secondary subclonal mutation(s), which could be responsible for a growth advantage; moreover, the PIG-A mutation could be a secondary event in the setting of an already established clonal expansion; finally, the PNH clone could expand in parallel with another mutated clone.
In our opinion, there is emerging evidence that even though the PIG-A mutation strongly influence the clinical manifestations, delineating the peculiar PNH phenotype, the PNH pathogenesis is probably extremely heterogeneous. This is in agreement with the fact that a PNH population can develop in different clinical settings (cytopenic or not), probably due to genomic instability or as an attempt to escape from an abnormal hematopoiesis, and that the evolution of the PNH clone (stabilization, remission or leukemic transformation) is extremely various and mostly unpredictable.
The rate of association between MPN and PNH has never been thoroughly investigated. According to the most accepted etiopathogenetic theory for PNH, this association is poorly explicable, but emerging evidence is demonstrating that this disease could be more heterogeneous than we thought. Our hypothesis is that some patients affected by MPN who experience important thrombotic events could carry an otherwise silent PNH clone, that could increase their thrombotic risk. In order to verify our hypothesis, we are currently designing a clinical trial where all MPN patients with recurrent or atypical thrombosis will be tested for the presence of a PNH clone.
PS, AC and VG contributed to the conception and design of the case presentation. MG, NS, FC and PB performed the flow cytometry and the molecular experiments. VG collected the data. VG, MG, AC and PS analyzed and interpreted the data. VG drafted the article. VG, AC, MG, PS, GS revised the manuscript. All authors read and approved the final manuscript.
Written informed consent was obtained from the patients for publication of this case report.
The authors declare that they have no competing interests.
- Hill A, Kelly RJ, Hillmen P (2013) Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood 121: 4985-4996. [Crossref]
- Young NS, Calado RT, Scheinberg P (2006) Current concepts in the pathophysiology and treatment of aplastic anemia. Blood 108: 2509-2519. [Crossref]
- Sugimori C, Padron E, Caceres G, Shain K, Sokol L, et al. (2012) Paroxysmal nocturnal hemoglobinuria and concurrent JAK2V617F mutation. Blood Cancer J 2: e63.
- Fraiman YS, Cuka N, Batista D, Vuica-Ross M, Moliterno AR (2016) Development of paroxysmal nocturnal hemoglobinuria in CALR-positive myeloproliferative neoplasm. J Blood Med 7: 107.
- Shen W, Clemente MJ, Hosono N, Yoshida K, Przychodzen B, et al. (2014) Deep sequencing reveals stepwise mutation acquisition in paroxysmal nocturnal hemoglobinuria. J Clin Invest 124: 4529-4538.
- Borowitz MJ, Craig FE, Digiuseppe J A, Illingworth AJ, Rosse W, et al. (2012) Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom 78: 211-230.
- Sutherland DR, Keeney M, Illingworth A (2012) Practical guidelines for the high-sensitivity detection and monitoring of paroxysmal nocturnal hemoglobinuria clones by flow cytometry. Cytometry B Clin Cytom 82: 195-208.
- Morado M, Freire Sandes A, Colado E, Subirá D, Isusi P, et al. (2016) Diagnostic screening of paroxysmal nocturnal hemoglobinuria: Prospective multicentric evaluation of the current medical indications. Cytometry B Clin Cytom.
- Lazo-Langner A, Kovacs MJ, Hedley B, Al-Ani F, Keeney M, et al. (2015) Screening of patients with idiopathic venous thromboembolism for paroxysmal nocturnal hemoglobinuria clones. Thromb Res 135: 1107-1109.
- http://www.netflowconnect.com/diagnosing-pnh/
- Primignani M, Barosi G, Bergamaschi G, Gianelli U, Fabris F, et al. (2006) Role of the JAK2 mutation in the diagnosis of chronic myeloproliferative disorders in splanchnic vein thrombosis. Hepatology 44: 1528-1534.
- Barcellini W, Fattizzo B (2015) Clinical applications of hemolytic markers in the differential diagnosis and management of hemolytic anemia. Dis Markers: 1-8.
- Beer PA, Campbell PJ, Green AR (2010) Comparison of different criteria for the diagnosis of primary myelofibrosis reveals limited clinical utility for measurement of serum lactate dehydrogenase. Haematologica 95: 1960-1963.
- Haan LD, Werre JM, Ruben AM, Huls AH, Gier J, et al. (1988) Reticulocyte crisis after splenectomy: evidence for delayed red cell maturation? Eur J Haematol 41: 74-79.
- Körmöczi GF, Säemann MD, Buchta C, Peck-Radosavljevic M, Mayr WR, et al. (2006) Influence of clinical factors on the haemolysis marker haptoglobin. Eur J Clin Invest 36: 202-209. [Crossref]
- Elliott MA, Tefferi A (2005) Thrombosis and haemorrhage in polycythaemia vera and essential thrombocythaemia. Br J Haematol 128: 275-290. [Crossref]
- Gruppo Italiano Studio Policitemia (1995) Polycythemia Vera: The Natural History of 1213 Patients Followed for 20 Years. Ann Intern Med 123: 656-664.
- Cortelazzo S, Viero P, Finazzi G, D'Emilio A, Rodeghiero F, et al. (1990) Incidence and risk factors for thrombotic complications in a historical cohort of 100 patients with essential thrombocythemia. J Clin Oncol 8: 556-562.
- Passamonti F, Rumi E, Pungolino E, Malabarba L, Bertazzoni P, et al. (2004) Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 117: 755-761. [Crossref]
- Hall C, Richards S, Hillmen P (2003) Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH). Blood 102: 3587-3591.
- Lee JW, Jang JH, Kim JS, Yoon SS, Lee JH, et al. (2013) Clinical signs and symptoms associated with increased risk for thrombosis in patients with paroxysmal nocturnal hemoglobinuria from a Korean Registry. Int J Hematol 97: 749-757.
- de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, et al. (2008) French Society of Hematology; French Association of Young Hematologists: Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood 112: 3099-3106.
- Hillmen P, Muus P, Dührsen U, Risitano AM, Schubert J, et al. (2007) Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood 110: 4123-4128.
- Samuelson BT, Vesely SK, Chai-Adisaksopha C, Scott BL, Crowther M, et al. (2016) The impact of ruxolitinib on thrombosis in patients with polycythemia vera and myelofibrosis: a meta-analysis. Blood Coagul Fibrinolysis 27: 648-652. [Crossref]
- Karadimitris A, Luzzatto L (2001) The cellular pathogenesis of paroxysmal nocturnal haemoglobinuria. Leukemia 5: 1148–1152.
- Shichishima T, Noji H (2002) A new aspect of the molecular pathogenesis of paroxysmal nocturnal hemoglobinuria. Hematology 7: 211-227.
- Gargiulo L, Papaioannou M, Sica M, Talini G, Chaidos A, et al. (2013) Glycosylphosphatidylinositol-specific, CD1d-restricted T cells in paroxysmal nocturnal hemoglobinuria. Blood 121: 2753-2761.