Abstract
Breast cancer is a heterogeneous disease. Hormonal blockade is the cornerstone of clinical management of the hormone-positive subtype; however, various mechanisms of resistance arise. One powerful mechanism of resistance is FGFR1 amplification. So far, pilot trials with in the advanced disease setting with FGFR1 inhibitors in breast cancer have yielded confounding results. FGFR1 amplification has been linked to hormonal resistance and increased relapse-rate after loco-regional treatment. With the current available data, there is high uncertainty regarding whether a large and expensive clinical trial in the adjuvant setting should be conducted. This review focuses on the positive and negative aspects of phase 0 clinical trials in order to make expedite go/no-go decisions for drug candidates in this area of research based on pharmacodynamic and pharmacokinetic parameters. We describe the key dose-escalation, pharmacodynamic and pharmacokinetic design aspects of the CNIO-BR-007 phase 0 trial, which combines the multikinase inhibitor nintedanib (a tyrosine-kinase inhibitor against VEGFR1-3, FGFR1-3 and PDGFR) with letrozole, a trial that aims to reach a definitive answer regarding whether or not nintedanib deserves testing in a large, randomized clinical trial in the adjuvant setting.
Key words
breast cancer, FGFR1, phase 0, nintedanib
FGFR and breast cancer
A major problem in breast cancer management is disease heterogeneity. Although the measurement of estrogen receptor, progestagen receptor, Ki67 fraction and HER2 receptor is able to classify the disease into 4 major clinical subtypes (hormone-receptor positive with high- or low-replication rate, HER2 positive or triple-negative breast cancer) [1], and each of these subtypes relies on a major specific therapeutic milestone (hormone blockade, chemotherapy, anti-HER2 antibodies and platinum-based chemotherapy, respectively), considerably differences are found in disease course, response to therapy and disease relapse after locoregional treatment.
Hormone-positive breast cancer accounts for approximately 2/3 of the incident cases. This is a particularly heterogeneous subtype [2]. Despite being hormone-receptor positive, patients usually present with rather different genomic landscapes and copy-number alterations [2,3]. It is not surprising that up to a dozen of different mechanisms of resistance against hormonal inhibitors have been described [4]. One of these mechanisms is the amplification of the fibroblastic growth factor receptor 1 (FGFR1) [5]. FGFR1 is a membrane-bound, tyrosine-kinase receptor that can activate and enhance signal transduction through the MAPK and Pi3K-AKT pathways upon ligand-binding [6]. Other FGFR family isoforms have been clearly linked to the development and progression of several malignancies, such as FGFR2 and FGFR3 activating mutations and urothelial or biliary tract carcinomas [6]. In these cases, selective pan-FGFR inhibitors have demonstrated impressive antitumor responses [7,8]. In breast cancer, FGFR1 amplification has been linked to increased relapse risk after loco-regional therapy, particularly in the hormone-positive subtypes [2,9]. However, its therapeutic role is less clear.
FGFR inhibitors in preclinical models and in advanced breast cancer
First-generation tyrosine-kinase inhibitors with anti-FGFRs activity were tested in advanced breast cancer, both in unselected patients and in FGFR1-amplified breast cancer [10,11]. In all cases, both toxicity and efficacy were at least and at best, respectively, moderate. The reason of these results is double: first, non-selective FGFR-inhibitors like dovitinib although they show FGFR1-inhibitory activity in vitro they have greater affinity for other receptors (such as VEGFR1-4, PDGFRA/B, KIT or FLT3). Thus, at tolerable doses they show important toxicity derived from their off-target activity and probably the FGFR1 inhibitory is sub-therapeutic. Second, preclinical experiments suggest that FGFR1, rather than a pure oncogenic-addiction driver, is a factor implicated in tumour progression, enhancing several virulent features such as hormonal resistance, metastatic potential or replication rate [9]. A synergistic effect with hormonal blockade has been shown both in vitro and in vivo in FGFR1-amplified breast cancer models [12]. Turner and Ashworth have shown that in FGFR1-amplified hormone-positive cancer cells, estrogen-receptor signalling is increased thanks to trans-activation of the Pi3K-AKT and MAPK pathways; in addition, these cells show baseline ligand-independent estrogen signalling. These changes are linked to resistance to inhibitors like tamoxifen. Arteaga and colleagues have also shown that FGFR1 alone is able to induce transcriptional activation of estrogen receptor-targeted genes even in absence of estrogens. Interestingly, it has been shown that the cross-talk between different receptors may condition the activity of different FGFR inhibitors: Turner and colleagues have shown in preclinical models that a high FGFR2-to-FGFR1 ratio conditions a mainly Pi3K-AKT signalling, compared to a mainly MAPK-signalling induced by the opposite ratio. The high FGFR2-to-FGFR1 ratio seems to be associated to high improved responses to FGFR inhibitors, however, when this occurs, FGFR2 is also able to trans-activate other tyrosine-kinase receptors. Considering that FGFR1 and FGFR2 can serve as a back-up signalling loop that compensate and sustain angiogenesis in presence of VEGFR1/2 tyrosine-kinase inhibitors, and taking the mentioned data together, it seems wise to combine hormonal blockade with multi-kinase inhibitors that block all the FGFR receptor family and if possible other membrane-bound tyrosine-kinase receptors in the clinical setting.
Nintedanib is a tyrosine-kinase inhibitor with highly promiscuous activity [13]. Nintedanib in vitro activity suggests highly selective activity for VEGFR2 and PDGFRB [13]. Because of this activity it is not surprising that it exerts a powerful antiangiogenic effect, correcting tissue hypoxia in animal models [14]. Non-invasive hypoxia tracing with 18F-fluoromisonidazole PET [14,15] has confirmed this stromal-normalizing effect in patients [16]. Nintedanib has demonstrated important clinical activity in lung and HER2-negative breast cancer [16-18]. Interestingly, nintedanib has a Km against FGFR1 of 69 nM [13], suggesting clinical activity at standard doses.
Is it worth launching a trial in the adjuvant setting with FGFR1 inhibitors? The importance of pharmacodynamics and pharmacokinetics
Currently, the adverse prognostic impact of FGFR1 amplification has been demonstrated in the adjuvant stage of the disease [9]. Addressing the role of nintedanib in this setting would require a placebo-controlled clinical trial in hundreds (if not thousands) of patients, with molecular selection of FGFR1 amplifications, and several years of follow-up. Such a trial would cost greater than 100 million USD and could also expose patients to unnecessary toxicity in case no activity is observed (which is, obviously, not warranted).
A phase-0/1 design allows at least to “shortcut” go/no-go decisions for candidate drugs in this setting. Two features would be an absolute requirement for this agent to demonstrate clinical benefit in the adjuvant state: 1) since the currently accepted recommended phase-II dose (RP2D) is optimized for the antiangiogenic effect, it is mandatory to demonstrate target engagement of FGFR1 at this dose level; 2) toxicity in combination with an hormonal agent must be, at most, moderate. Phase 0 trials can expedite the subsequent development of a promising agent by providing essential pharmacodynamic and pharmacokinetic data. Regarding the potential disadvantages or complexities of phase 0 trials, they require validated assays for pharmacodynamics and standardized tissue handling procedures.
A phase 0/I trial in order to make a go/no-go decision
The CNIO-BR-007 trial is an investigator-initiated, multicentric, phase 0/phase I trial that studies the safety, pharmacokinetics and pharmacodynamics of the combination of letrozole and nintedanib. The objectives are: 1) to define the recommended dose for phase II; 2) to study the interactions between nintedanib and letrozole pharmacokinetics; and 3) to ensure FGFR1 engagement by nintedanib at the recommended dose for phase II. Figure 1 depicts the basic trial schedule. Patients that have been adequately treated for any early hormone-receptor positive breast cancer (any T, any N, ER and/or PR>5%, any grade and Ki67 values, that have received surgery and/or chemotherapy and/or radiation therapy according to NCCN guidelines) and have been on adjuvant letrozole for less than 6 months are candidates to enter the trial. Patients are recruited with no active or measurable disease – thus, efficacy is not an endpoint of this trial. However, demonstrating pharmacodynamic activity, tolerability, and lack of significant pharmacokinetic interactions (i.e., achieving the three main objectives of the trial) are necessary conditions to obtain efficacy in the future. Patients are only on trial for one month. Within this 4-week period, the patients undergo a full nintedanib- and letrozole-pharmacokinetic profile (baseline and steady-state, at week 4; both times patients are drawn a blood sample before and 15min, 30min, 1h, 2h, 4h, 8h and 24h after intake of letrozole and nintedanib). It has been demonstrated that blocking FGFR1 signalling in the bone induces two compensatory effects: 1) increase in free phosphate19; 2) increase in FGF2319. Thus, baseline, after 2 weeks and after 4 weeks both FGF23 and phosphate are determined in plasma. Regarding nintedanib dosing, although letrozole dose is fixed at the standard dose (2.5 mg daily), nintedanib is escalated starting at 150 mg BID and patients are enrolled in this trial following a classic 3+3 dose-escalation schedule. Once the RP2D is determined, 10 additional patients will be recruited at that dose-level, in order to increase the number of patients with full pharmacokinetic and pharmacodynamic data. Finally, after completing the 4-weeks course, patients are offered to continue on trial until a total period of 6 months, given the observed positive benefits of nintedanib in early breast cancer. By doing this, we will also gather data regarding long-term tolerability. Although long-term toxicities are not commonly reported in phase I trials, it is important to learn about the incidence of persistent grade 2 “inconvenient” toxicities such as diarrhea, rash or hand-foot syndrome. These toxicities rarely cause treatment interruption within the first month, and usually are not highly relevant in short treatment courses as those in metastatic patients, who are usually willing to tolerate greater toxicities. However, for a potential future trial in the adjuvant setting, where therapies are administered for a year or longer in fit patients with – most of the times – active lifestyles, it will be highly relevant to know beforehand the drop-out rate after prolonged drug exposure and potential extended incidence of low-grade toxic events.
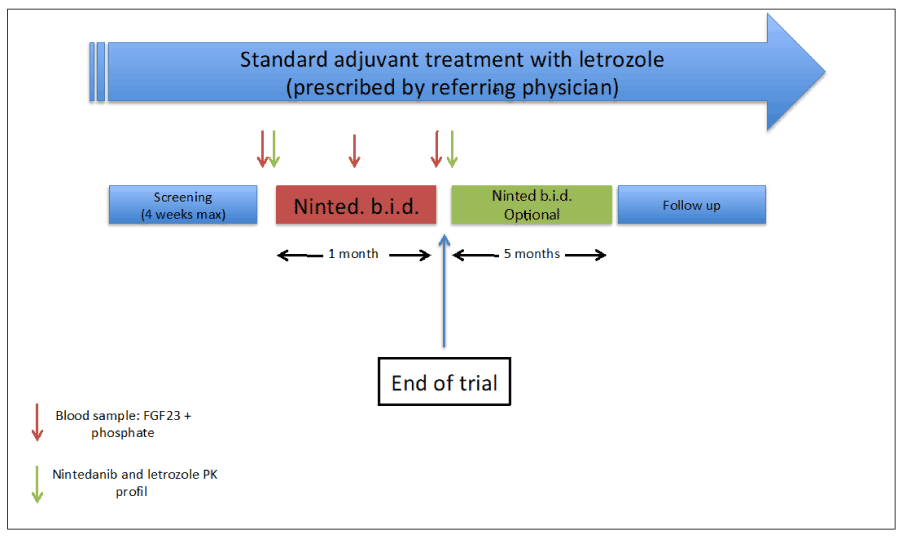
Figure 1. CNIO-BR-007 design: Patients adequately treated for non-metastatic hormone-positive breast cancer receiving adjuvant letrozole for less than six months are candidate for this trial. Patients start on Nintedanib (level 1: 150 mg BID) in day one, and undergo a full pharmacokinetic profile (samples are drawn before drug intake and then after 15 min, 30 min, 1hour, 2 hours, 4 hours, 8 hours and 24 hours). The pharmacokinetic profile is repeated after 4 weeks of treatment. Pharmacodynamics is assessed by serial determination of blood phosphate and FGF23 levels: baseline, after 2 weeks of combined treatment and after 4 weeks. Depending on toxicity and patient’s decision, they can continue for 5 additional months on combination therapy.
Conclusion
Considering that only 5% of the agents that enter phase I achieve registration, it is imperative to afford costs and resources as well as to decrease the exposure of patients to potentially inactive while toxic agents. Better preclinical models and the integration of preclinical models and TCGA data will allow increasing the approval rate; however, in order to optimize the use of compounds and the expense of resources, phase 0 trials are a powerful tool that can help adopting go/no-go decisions. Our CNIO-BR-007 trial hopefully will answer weather nintedanib, an agent with in vitro activity against FGFR, warrants clinical testing in the adjuvant setting as it will demonstrate whether at long-term tolerable doses is able to block FGFR1 without interfering with the standard of care hormonal treatment.
References
- Cheang MC, Chia SK, Voduc D, Gao D, Leung S, et al. (2009) Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 101: 736-750. [Crossref]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, et al. (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486: 346-352. [Crossref]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, et al. (2012) Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486: 353-360. [Crossref]
- Ma CX, Reinert T, Chmielewska I, Ellis MJ (2015) Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer 15: 261-275. [Crossref]
- Johnston SR (2015) Enhancing Endocrine Therapy for Hormone Receptor-Positive Advanced Breast Cancer: Cotargeting Signaling Pathways. J Natl Cancer Inst 107. [Crossref]
- Touat M, Ileana E, Postel-Vinay S, André F, Soria JC (2015) Targeting FGFR Signaling in Cancer. Clin Cancer Res 21: 2684-2694. [Crossref]
- Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, et al. (2017) Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov 7: 252-263. [Crossref]
- Pearson A, Smyth E, Babina IS, Herrera-Abreu MT, Tarazona N, et al. (2016) High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov 6: 838-851. [Crossref]
- Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, et al. (2010) FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 70: 2085-2094. [Crossref]
- André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, et al. (2013) Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 19: 3693-3702. [Crossref]
- Musolino A, Campone M, Neven P, Denduluri N, Barrios CH, et al. (2017) Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR+, HER2- breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res 191: 18. [Crossref]
- Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero-Zotano AL, et al. (2017) Association of FGFR1 with ERalpha Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER(+) Breast Cancer. Clin Cancer Res 23: 6138-6150. [Crossref]
- Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, et al. (2008) BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res 68: 4774-4782. [Crossref]
- Navarro P, Bueno MJ, Zagorac I, Mondejar T, Sanchez J, et al. (2016) Targeting Tumor Mitochondrial Metabolism Overcomes Resistance to Antiangiogenics. Cell Rep 15: 2705-2718. [Crossref]
- Hernandez-Agudo E, Mondejar T, Soto-Montenegro ML, Megias D, Mouron S, et al. (2016) Monitoring vascular normalization induced by antiangiogenic treatment with (18)F-fluoromisonidazole-PET. Mol Oncol 10: 704-718. [Crossref]
- Quintela-Fandino M, Lluch A, Manso L, Calvo I, Cortes J, et al. (2017) 18F-fluoromisonidazole PET and Activity of Neoadjuvant Nintedanib in Early HER2-Negative Breast Cancer: A Window-of-Opportunity Randomized Trial. Clin Cancer Res 23: 1432-1441. [Crossref]
- Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, et al. (2014) Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol 15: 143-155. [Crossref]
- Quintela-Fandino M, Urruticoechea A, Guerra J, Gil M, Gonzalez-Martin A, et al. (2014) Phase I clinical trial of nintedanib plus paclitaxel in early HER-2-negative breast cancer (CNIO-BR-01-2010/GEICAM-2010-10 study). Br J Cancer 111: 1060-1064. [Crossref]
- Wohrle S, Bonny O, Beluch N, Gaulis S, Stamm C, et al. (2011) FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J Bone Miner Res 26: 2486-2497. [Crossref]