Ten years ago, in July 2004, a “medical mystery” involving 50 year old homozygot twins was presented in NEJM [1]. In an August issue the solution of this mystery showed that one of the twins had acromegaly.
We here report a singular case where “mystery” resides in the succession of seeming contradiction, this time in the same person.
At presentation, this girl showed retarded growth (growth curve figure 1), with rupture in growth speed at the age of 11. Diagnosis of GH deficiency was based on three stimulation tests (Insulin-Arginin, Ornicetin, Clonidine-TRH) on GH. Two of them showed insufficient response and one was normal. Treatment by recombinant human Growth Hormone (hGH) was followed during a bit more than a year and a half (July, 1995 until February, 1997), between the age of 13years 11 months and 15 and a half). The prescribed dose was between 0,47 to 0,60 IU/kg/week. Follow-up was stopped after end of treatment, height had reached 160 cm. During following years, growth kept on until final height of 167 cm, reached at the age of 20, unusually late in females. Let us notice that, at the time, hGH therapy was changed, and changing in its modes, prescription responsibility, and decision being transferred from “France Hypophyse”, a national centralizing organism, to endocrinologists (under their own responsibility); In our case, decision was endorsed and follow-up of hGH treatment was taken on by a teaching hospital. This doesn’t avoid to think that diagnosis of GH deficiency wasn’t so sure, and that a delayed puberty was likely. Bone age evaluation at the age of 13 ½ was 12 ½. At the age of 13, plasmatic Oestradiol was below 1 ng/dl, LH and FSH were below limits of analysis, pelvic ultrasound showed prepubertal uterus and ovaries. Caryotype was normal, 46 XX. Let us notice that first periods appeared at the age of 15.
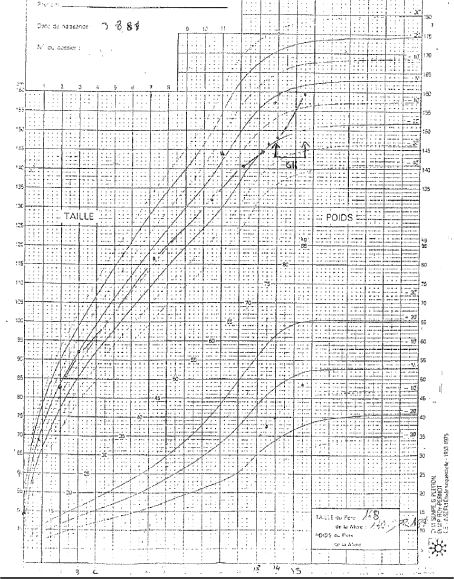
Figure 1. Courbe Croissance (Growth Curve)
Seven to eight years went by.Headaches appeared, worsening, accompanied by disorders of menstrual periods (10 month amenorrhea). Within some months, by the end of 2010, it led to diagnosis of acromegaly on elevated IGF-1 and lack of GH suppression during glucose tolerance test. A moderate hyperprolactinemia was also discovered (29 ng/ml). The MRI showed macroadenoma (Figures 2 and 3). After a period of medical treatment with somatostatin analogs and dopaminergic (quinagolide 75 micrograms per day), trans-sphenoidal surgery was decided, GH secretion being unchecked, and because of proximity between chiasma and the tumor. Debulking was successfully carried out on March the 9th of 2012. Thyrotroph and corticotroph deficiencies were discovered, and treated was started. GH secretion was checked during a while (three months), with somatostatin analog (SMS analog, Sandostatin® late release, 30 mg every four then three weeks) and addition of pegvisomant (Somavert®) daily (10 mg then 20 mg per day), for July 2012. Some problems of observance with pegvisomant were noticed, as classically, due to lipodystrophy appearance [2].
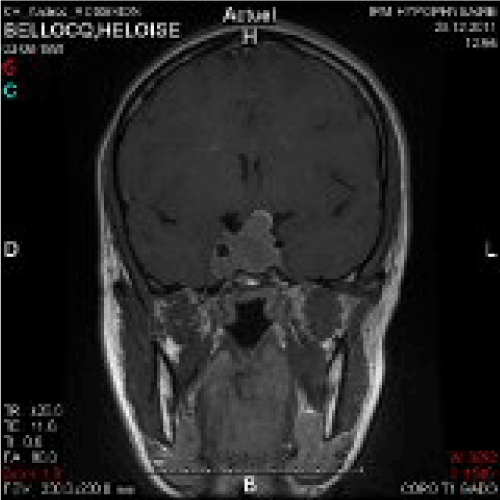
Figure 2. MRI 1
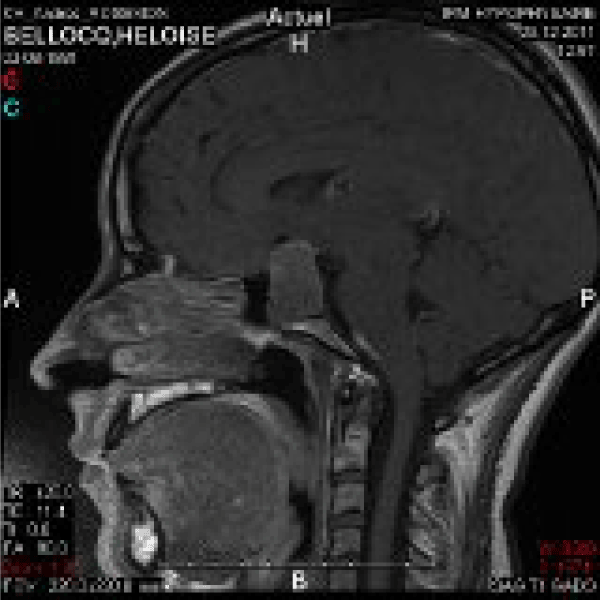
Figure 3. MRI 2
Notwithstanding, Radiotherapy (proton-therapy, from December, 2013 to January, 2014) proved to be necessary, with an increase in IGF-1 despite pegvisomant and SMS analog at maximal doses. Gamma-knife [3] and conventional radiotherapy were discussed, in order to preserve gonadal function, however active in approximately one third of acromegalic female patients during fertility duration [4].
The patient is now treated by Somavert® 20 mg per day, Sandostatin® 30 mg every three weeks, Hydrocortisone® 20 mg per day, Thyroxine (levothyrox®) 100 micrograms per day,
Remarkably, the patient has a sister, treated also by h GH during teenage years.
Nevertheless, sequencing of AIP and menin genes did not find any mutation, as in the vast majority of cases [5].
To our knowledge, this is the 1st description of treated Growth Hormone (GH) deficiency during teenage years, followed 13 years after by the diagnosis of acromegaly. GH deficit was partial during adolescence, somatotropic cells were present.
What one is allowed to think, is that the somatotrophic adenoma, made of a monoclonal proliferation, descended from one of those cells, appeared maybe just a few after initial evaluation. Indeed, continuation of growth after the age of 15 and a half, until 20, is an argument. There was about one year of delay on bone age evaluation at the age of 13 ½, and two years of delay at the age of 15 ½. This could partly explain why prolonged growth after usual end (in females) was able to occur in the case of this patient. However, delayed start and ending of puberty in this patient explains most of this late ending of growth.
Another question, to which this unusual situation leads, is of GH secretion stimulation tests are possibly impaired in acromegaly, and if GH slow-down tests are impaired in GH deficiency. We found no study on these subjects (Cochrane and Pubmed sources)
Searching for similar cases in bibliography (PubMed) didn’t show any previous cases of this sequence, GH “down and up”. The closest we found was a publication of Japanese colleagues [6], reporting the case of a girl, GH deficient (very low and non stimulable plasma values), and in the same time having excess height growth rate, and acromegaly symptoms (more precisely gigantoacromegaly). Of course, the only common point is acromegaly, but no similarity, rather opposite features about clinical presentation (growth delay-growth acceleration)
Apart from this so peculiar succession of this hormonal lack and excess, one can also notice the young age at onset of symptoms, and even diagnosis, of acromegaly in this patient. Indeed, peak incidence occurs in middle age, usually around forty years old [7,8]. Yet, our patient was around 25, when the first symptoms appeared.
Anyway, let’s not forget that one extreme situation in pathology does not exclude the opposite, in the same person, within just a few years. Pathologies characterised by alternation of deficiency and excess (or conversely) are well described for a long time. We can mention De Quervain’s subacute thyroiditis [9], and somehow also type 2 diabetes. This should remind us that a hormonal (or any other kind) deficit does not exclude the excess in the following story of patients, sometimes quite astonishing like there.
- Nieuwlaat WA, Pieters GA (2004) Medical Mystery Which Twin Is the Patient? N Engl J Med 351: 68.
- Sesmilo G, Resmini E, Bernabeu I, Aller J, Soto A, et al. (2014) Escape and lipodystrophy in acromegaly during pegvisomant therapy, a retrospective multicentre Spanish study. Clin Endocrinol (Oxf) 81: 883-90. [Crossref]
- Morange-Ramos I, Regis J, Dufour H, Andrieu JM, Grisoli F, et al. (1998) Gamma-knife surgery for secreting pituitary adenomas. Acta Neurochir (Wien) 140: 437-443. [CrossRef]
- Grynberg M, Salenave S, Young J (2010) Female gonadal function before and after treatment of acromegaly. J Clin Endocrinol Metab 95: 4518-25. [Crossref]
- Zhou Y, Zhang X, Klibanski A (2014) Genetic and epigenetic mutations of tumor suppressive genes in sporadic pituitary adenoma. Mol Cell Endocrinol 386: 16-33. [CrossRef]
2021 Copyright OAT. All rights reserv
- Iwatani N, Kodama M, Miike T (1992) Endocrinological evaluation of GH deficient patient with acromegaloidism showing excessive growth. Endocrinol Jpn 39: 59-64. [Crossref]
- Sesmilo G (2013) [Epidemiology of acromegaly in Spain]. Endocrinol Nutr 60: 470-474. [CrossRef]
- Sala E, Ferrante E, Locatelli M, Rampini P, Mantovani G, et al. (2014) Diagnostic features and outcome of surgical therapy of acromegalic patients: Experience of the last three decades. Hormones 13: 95-103. [Crossref]
- Slatosky J, Shipton B, Wahba H (2000) Thyroiditis: differential diagnosis and management. Am Fam Physician 61: 1047-1052, 1054. [CrossRef]