Human parasitic infections constitute a substantial but neglected disease burden. Protozoa and helminths are two parasites groups known to be pathogenic to the human heart. Trypanosoma cruzi, Trypanosoma brucei, toxoplasma, plasmodium and Leishmania are the most frequent protozoa that can establish infections in human resulting in a wide spectrum of cardiac manifestations. The involvement of the myocardium can manifest as protozoan cardiomyopathy in many resource-constrained countries. However, at present, resource-rich countries are encountering diagnoses of parasitic infections with cardiac involvement attributable to increasing travel and migration, blood transfusions, growing numbers of immunodeficient patients – a worldwide epidemic of acquired immunodeficiency syndrome (HIV/AIDS), and increasing transplant recipients and use of immunosuppressive agents. Clinicians across the globe need to be aware of the potential cardiac involvement after parasitic infection in vulnerable individuals and in endemic areas. This paper reviews published evidence on protozoal parasites that can infect the myocardium, including the pathophysiology, diagnosis and management of protozoan cardiomyopathy. The paper also highlights areas of limited knowledge that could benefit from additional research.
cardiomyopathy, parasitic, protozoal, chagas disease
Abbreviations: ACE-I: Angiotensin Converting Enzymes – Inhibitors; AIDS: Acquired Immunodeficiency Syndrome; AV: Atrio-Ventricular; CAD: Coronary Artery Disease; CAF: Cerebro-Spinal Fluid; CATT: Card Agglutination Trypanosomiasis Test; CM: Cardiomyopathy; DCM: Dilated Cardiomyopathy; ECG: Electrocardiograph; Echo: Echocardiography; ELISA: Enzyme-Linked Immunosorbent Assay; EMB: Endomyocardial Biopsy; ESC: European Society of Cardiology; HAT: Human African Trypanosomiasis; HF: Heart Failure; HIV : Human Immuno-Deficiency Virus; ICD: Implantable Cardioverter Defibrillator; IFN-γ: Interferon Gamma; IgG: Immunoglobulin G; IIF: Indirect Immunofluorescence; LAFB: Left Anterior Fascicular Block; LV: Left Ventricular; LVEF: Left Ventricular Ejection Fraction; NT-pro-BNP: N-terminal-prohormone Brain Natriuretic Peptide; NYHA: New York Heart Association; PCM: Protozoan Cardiomyopathy; PCR: Polymerase Chain Reaction; PVC: Premature Ventricular Contractions; RBBB: Right Bundle Branch Clock; RCM: Restrictive Cardiomyopathy; RV: Right Ventricular; VT: Ventricular Tachycardia; WHO: World Health Organization
Historically, the epidemiologic pattern of cardiac diseases varied between resource-constrained and resource-rich countries [1-4]. In 2008, the World Health Organization (WHO) estimated that 75% of the world’s burden of cardiac diseases would be in resource-constrained countries [5]. At present, the importance of infectious aetiologies of cardiac diseases in resource-constrained countries is becoming increasingly recognized [6]. However, cardiac manifestations previously only seen in resources constrained countries including certain protozoan infections have begun to feature in resource-rich countries [7-9]. This epidemiologic shift is attributable to (i) growing human travel and migration [10-12]; (ii) increasing blood transfusions; and (iii) growing numbers of immunodeficient individuals associated with the worldwide epidemic of acquired immunodeficiency syndrome (HIV/AIDS), and increasing transplant recipients and use of immunosuppressive agents [13-18]. Parasitic infection due to protozoa frequently has cardiac and pulmonary involvement. Cardiac involvement may be a part of a more generalized disease or a direct effect on various anatomic structures of the heart – myocardium, pericardium, endocardium or the cardiac vasculature [7,8]. The involvement of the myocardium may lead to myocarditis, or different types of cardiomyopathies – dilated or restrictive [9]. This systematic review and meta-analysis synthesizes current evidence on protozoal cardiomyopathy (PCM) including causative protozoal pathogens, pathophysiology, diagnosis and clinical management strategies of PCM.
Clinical definition
The 2008 position statement by the European Society of Cardiology (ESC) Working Group on Myocardial and Pericardial Diseases defines cardiomyopathy (CM) as “a myocardial disorder in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular disease and congenital heart disease sufficient to cause the observed myocardial abnormality” (p. 271) [19]. Persistence protozoan infection and presence in the myocardium may lead to structurally distinct forms of cardiomyopathy –more so dilated and restrictive cardiomyopathies. Dilated cardiomyopathy (DCM) is a spectrum of heterogeneous myocardial disorders characterized by left ventricular (LV) dilatation and LV systolic dysfunction in the absence of abnormal loading conditions (hypertension, valve disease) or coronary artery disease (CAD) sufficient to cause global systolic impairment [19,20]. Although right ventricular (RV) dilatation and dysfunction may be present, they are not necessary for diagnosis [19]. Restrictive cardiomyopathy (RCM) on the other hand is an uncommon myocardial disorder characterized by normal or reduced biventricular volumes associated with bi-atrial enlargement, normal LV wall thickness and atrioventricular (AV) valves, impaired ventricular filling with restrictive physiology, and normal or near normal systolic function [20].
Protozoal aetiologies
The causes of CM can be demonstrable (known) or unknown (idiopathic) [21]. The epidemiology of demonstrable causes vary across the world, with viruses frequently cited as the most common causes in Europe and North America, although worldwide, the most common cause may be due to protozoan infection, which is endemic to resource-constrained countries of Central and South America, and Africa [8,9]. Typically, protozoa are a diverse group of unicellular eukaryotic organisms but pathogenic protozoa, often described as parasites, can cause a wide array of clinical diseases. The most frequently implicated protozoa pathogens that infect the myocardium leading to PCM include Trypanosoma, Toxoplasma, Plasmodium, Entamoeba, Leishmania, Balantidium, and Sarcocystis.
American trypanosoma (Chagas Disease): Trypanosoma cruzi (also known as American Trypanosoma) is a flagellate protozoan parasite and the aetiologic agent of Chagas disease (also known as American trypanosomiasis). T. cruzi is endemic in South America, Central America and some parts of North America (Southern United States and Mexico). Historically, Chagas disease was predominant in resource-constrained settings because the transmission of T. cruzi infection mainly occurred in rural areas characterized by poor quality housing and close contact with potential vectors [22]. The most epidemiologically important vectors live in the cracks in mud walls and thatched roofs of rustic rural house leading to repeated exposure to vectors and parasites of rural inhabitants [23]. However, in the past five decades, there has been a marked shift in the epidemiology of Chagas disease, demonstrated in reports of diagnosis in both urban and peri-urban settings as well as in both endemic and non-endemic areas [22]. The epidemiologic shift is attributable to domestic vector-borne transmission over the lifetime of the current population of Latin America partnered with large-scale rural-urban and international migration [23-25].
The principal infection pathway is vector-borne transmission by traitomine insects in areas of endemicity although infection in non-endemic areas may also occur via blood transfusion, organ transplant or vertical transmission (from mother to child at birth) and less commonly through ingestion of contaminated food [9,26]. Traitomine insect transmits the infective form of T. cruzi (the metacyclic trypomastigotes) by biting an infected person or animal and defecating on the host skin or on the mucous membranes [26]. After infection, the trypomastigotes invade local host cells and differentiate into amastigotes. They multiply intracellularly causing the host cell to become swollen with amastigotes, which then transform back into trypomastigotes by growing flagella. The trypomastigotes lyse the cells, invade adjacent tissues and disseminate via the lymphatics and blood stream to cause systemic infection [23,24]. The resulting Chagas disease manifests in three phases: (i) acute; (ii) indeterminate; and (iii) chronic (or CM) [23-26]. (Figure 1).
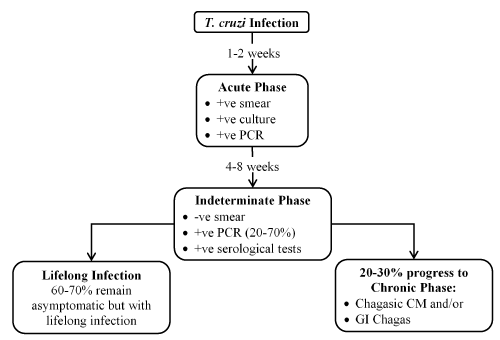
Figure 1. Clinical phases of chagasic cardiomyopathy
In a minority of patients, the initial infection (1-2 weeks) is characterized by the acute phase, which is often asymptomatic and may manifest as a self-limiting febrile illness lasting about 4 to 8 weeks [23,26]. About 60% to 70% of these patients will not develop clinically apparent disease but remain healthy and asymptomatic with lifelong infection. This second phase is the indeterminate form of Chagas disease, characterized by positive serological tests but with no demonstrable alterations of the heart, oesophagus and colon [26]. However, these patients exhibit persistent infection with parasitemia that is demonstrable more than 20 years after the initial infection [27]. At some point, usually 10 to 30 years after the initial infection, some patients in the indeterminate phase may develop symptoms, entering the third phase of overt disease [28,29]. About 20% to 30% of infected individuals will develop chronic CM – described as the most serious manifestation of Chagas disease as well as the most common type of chronic myocarditis [30]. Definitionally, Chagasic CM encompasses all cases of Chagas disease with cardiac involvement, defined by the presence of at least one typical electrocardiographic (ECG) abnormality in patients who have positive serological tests for T. cruzi. Dilated Chagasic CM refers to the hemodynamic pattern of Chagasic CM characterised by LV enlargement with segmental or global systolic dysfunction despite electrographic findings [31].
Pathophysiology: The 2018 scientific statement from the American Heart Association (AHA) on Chagasic CM provides valuable updates on the current knowledge on pathogenic and pathophysiologic mechanisms of Chagasic CM [31]. Although the pathophysiology of Chagasic CM is complex and incompletely understood, current evidence suggest two primary and two secondary mechanisms [31,32]. The primary mechanisms are (i) parasite-driven immune response; and (ii) auto-reactivity triggered by the infection to initiate and drive both acute and chronic myocardial inflammation. The two secondary mechanisms are (i) neurogenic disturbances and coronary microvasculature derangements responsible for cardiac alterations [33-35]. The pathogenic differences in T. cruzi strains and host susceptibility may likely play a role in different clinical patterns and disease severity [36].
In the initial acute phase of infection, cardiac inflammation and damage may be the consequence of high-grade parasitemia and intense direct tissue parasitism [31]. Although historically debatable, the development of more sensitive modern diagnostic techniques demonstrate that chronic Chagasic heart disease is an infectious CM characterized by incessant low-grade inflammation due to continued presence of T. cruzi in cardiac tissues [37-43]. Accumulating evidence supports the pathogenic role of tissue parasitism and immune response. Post-mortem studies and myocardial biopsies show a close correlation between T. cruzi tissue burden, and the site and intensity of the inflammatory processes [44-46]. Animal models and human studies show a correlation between the severity and distribution of MC as well as the severity and pattern of heart failure (HF) [47,48]. Experimental animal models show a correlation between host parasite burden and severity of cardiac inflammation [49-54]. Finally, the presence of apoptosis in the advanced JF stages suggests direct parasitic myocyte aggression contributes to ongoing inflammation [55].
Cellular and possibly humoral immune responses targeting parasitic infection may eventually control the initial acute infection but may fail to completely eliminate the parasite. A variably long asymptomatic (indeterminate) phase ensues. In this phase, parasite strain, parasite load during the prior acute phase, the quality of host immune response and the presence or absence of re-infection may influence the course of the chronic disease [56,57]. Patients in the indeterminate phase often exhibit a balanced production of inflammatory and anti-inflammatory cytokines but those who go on to develop CM and HF appear to lose this co-regulation [31]. The pathophysiologic mechanisms underlying the characteristics myocardial tissue damage in the chronic phase of Chagasic CM is not clear although a combination of the direct immune response targeting parasite infection and infection-triggered autoreactivity may likely produce significant ongoing inflammation, myocytolosis and super-imposed reactive and reparative fibrosis [58-64].
Neurogenic disturbances and coronary microvascular derangements are two possible secondary mechanisms playing an important role in cardiac alterations in patients with Chagasic CM [31]. Parasympathetic neuronal depopulation is a general characteristic of CM. However, in Chagasic CM, neuronal depopulation in the heart, oesophagus, and colon of both humans and animal models is more severe and extensive [65-69]. In a majority of individuals with Chagas disease, the loss of parasympathetic cardiac control manifests prior to the onset of myocardial dysfunction [70-74]. Despite the parasympathicopriva Chagas cardiopathy theory postulating that catecholamine-induced CM is a consequence of parasympathetic dysfunction [75,76], it does not explain the lack of correlation between considerable individual variability of vagal denervation with severity of LV dysfunction and its detection in many patients. However, the theory is useful in explaining impaired control of the coronary microcirculation that results from dysutonomia [77,78]. Parasympathetic impairment may also be responsible for triggering malignant arrhythmia and sudden death evidence in pathological reports of highly denervated hearts of Chagasic patients with sudden death [65,79]. Nevertheless, the pathogenesis of arrhythmia in Chagasic CM patients is possibly multifactorial, also including adrenergically denervated myocardium and abnormal neuro-immunomodulatory anti-inflammatory functions of the parasympathetic nervous system [76,80,81].
There is also evidence of microvascular derangements contributing to cardiac alterations in Chagasic CM patients. Direct effect of T. cruzi or autoimmune reaction may lead to vascular endothelial cell damage, and subsequently microvascular perfusion derangement, which may result in ischemia-like symptoms (Chagas chest pain syndrome), ST- and Q-wave changes, and impaired regional wall motion [82,83]. Abnormalities in coronary microvasculature including increased platelet activation, micro-thrombi, focal spasm and endothelial dysfunction have been described in patients and animal models [84,85]. Human studies evaluating cardiac biopsy and necroscopy specimens of patients infected with T. cruzi describe findings similar to those observed in transient microvascular ischemia [85,86]. The pathogenic role of microvasculature is also evident in studies showing myocardial perfusion abnormalities in patients with Chagas disease with angiographically normal coronary arteries and sequential perfusion scans correlating deteriorating LV systolic function with irreversible perfusion defects over time [87].
Clinical presentation: Chagasic CM represents the most clinically relevant manifestation of Chagas disease, which is responsible for a greater proportion of Chagas disease-associated morbidity and mortality [88]. The ESC [19] and the AHA [20] classification of CM categorize Chagas disease an aetiology of DCM based on its hemodynamic pattern. However, Chagasic CM has a typical predominant distribution of fibrosis to the posterior and apical regions of the LV and the involvement of the sinus node and electric conduction system distinguishing it from other cardiomyopathies [31]. Clinical manifestations of Chagasic CM results from impaired cardiac function [32]. Common clinical manifestations include electrical conduction abnormalities, myocardial contractile dysfunction, arrhythmias and/or thromboembolism, apical aneurysm or sudden cardiac death [9,11,20,31] (Table 1).
Table 1. Typical clinical presentation of chagasic cardiomyopathy
Presentation |
Types, organ affected, frequency or causes |
Arrhythmias |
Ventricular extra-systoles; non-sustained and sustained ventricular tachycardia; bradyarrhythmia; ventricular fibrillation; atrial fibrillation or flutter |
Conduction abnormalities |
Sick sinus syndrome; complete/incomplete right bundle branch block, left anterior fascicle block; bifascicular and trifascicular blocks; 1st,2nd and 3rd degree atrioventricular blocks |
Thromboembolic events |
Brain (most frequent); lungs, kidney, spleen |
Cardiac Failure |
Diastolic dysfunction (initially); isolated left heart failure in early stages of cardiac decompensation; bi-ventricular with a predominance of right-sided HF (advances stages) |
Apical aneurysm |
Found in 52% of autopsy series; more frequent in men; 80% in the LV apex |
Sudden cardiac death (causes) |
Ventricular fibrillation (most frequent); bradyarrhythmia; rapture of apical aneurysm (very rare) |
In asymptomatic or oligosymptomatic patients, the transition from indeterminate to chronic form of Chagas disease usually manifests clinically by the appearance of ECG abnormalities such as incomplete or complete right bundle branch clock (RBBB), left anterior fascicular block (LAFB), minimal ST-T changes, monomorphic premature ventricular contractions (PVC) [28,89]. As the disease progress, associated intraventricular conduction defects (RBBB with LAFB), polymorphic PVCs, bradyarrhythmia, high-grade AV blocks, Q waves, non-sustained or sustained ventricular tachycardia (VT) and ultimately atrial flutter or fibrillation may follow. Clinical symptoms may include palpitations, atypical chest pain, pre-syncope, exertional dyspnoea, oedema occur throughout the natural course of Chagasic CM [9]. Physical signs vary based on the stage of the disease and the presence of conduction abnormalities. Common signs include cardiac rhythm irregularities, displaced point of maximal impulse, gallop rhythms, loud second heart sound (suggesting pulmonary hypertension), mitral or tricuspid regurgitation murmurs, increased systemic venous pressure with liver enlargement and oedema, and borderline low systolic blood pressure with reduced radial pulp (suggesting systolic dysfunction) [28].
Echocardiography findings reveal diastolic dysfunction usually precedes systolic dysfunction potentially enabling early detection of Chagas disease [90]. Typical echocardiography findings include apical aneurysms in 8.5% to 55% of the cases depending on stage of the disease and method of detection (post-mortem, echocardiography or angiography), segmental LV contractile abnormalities and depressed LV systolic function [91]. Echocardiographic evidence of LV dysfunction include increase in LV systolic dimensions, reduced LV ejection fraction (LVEF) or the presence of segmental or global LV wall motion abnormality and/or LV aneurysm. Echocardiographic evidence of LV dysfunction is the most common and consistent independent predictor of death in patients with Chagasic CM [92]. Other important clinical and non-invasive ominous prognostic indicators suggesting the extent of myocardial dysfunction include New York Heart Association (NYHA) functional class III/IV, cardiomegaly and non-sustained VT on 24-h ECG monitoring [92].
Despite ECG and echocardiographic changes, the diversity in the natural course of the Chagasic CM to complicate the ability to predict patients that will develop the disease or not. Some patients remain asymptomatic for life despite ECG and/or echocardiographic evidence of the disease, some present with signs and symptoms, and complications of progressive HF or severe cardiac arrhythmias while other die suddenly without prior clinical symptoms [92]. To assist clinicians to identify patients at different degrees of risk, facilitate treatment choices and assist in patients counselling, several staging systems are available [9]. Most of these systems classify patients based on four or five stages depending on their functional capacity, ECG findings and the presence or absent of cardiomegaly and/or echocardiographic defined systolic dysfunction [9] (Table 2). Staging is also important for prognostication. Mortality rate of patients at early stages of Chagasic CM is comparable to that of the general population while survival is < 30% at five years for symptomatic patients at advanced stages (those with systolic dysfunction and/or cardiomegaly) and their prognosis is more ominous compared to non-Chagas DCM aetiologies [93-95].
Table 2. Staging system for cardiac involvement in chagas disease
Stage |
Description (Functional Capacity, ECG, Echo, Cardiomegaly) |
1st |
Indeterminate phase (No cardiac involvement demonstrated by)
ECG and CXR (stage 0 KC) [96]
ECG, echo and signs of CHF (stage IA MLAC) [97]
ECG, echo, CXR and NYHA (stage A-ACC/AHA) [98,99] |
2nd |
Chagasic CM without signs/symptoms of heart failure
Evidence of structural heart disease demonstrated by:
ECG ± CXR (stage I-II KC) [96]
ECG ± echo (stage A-B2 BCC) [100]
Echo ± ECG (stage IB-II MLAC) [97]
ECG (stage B A-ACC/AHA) [98,99] |
3rd |
Compensated Chagasic CM (Considers symptoms)
Compensated CHF (stage C BCC) [100].
NYHA II-III (stage c A-ACC/AHA) [98,99] |
4th |
Overt, refractory or advanced Chagasic CM
Stage III from the KC and the MLAC [96,97]
Stage D from the BCC and the A-ACC/AHA [98-100] |
ACC/AHA: American College of Cardiology/American Heart Association; BCC: Brazilian Consensus Classification; CHF: Congestive Heart Failure; CXR: Chest Radiograph; KC: Kuschnir classification; MLAC: Modified Los Andes Classification; NYHA: New York Heart Association
Systemic and pulmonary embolism resulting from mural thrombi in the cardiac chambers are relatively frequent. The brain is the most common clinically recognized site of embolism followed by limbs and the lungs but at necropsy, lungs, kidneys and spleen are frequent sites for embolism [101]. In endemic areas, Chagas disease is an independent risk factor for stroke [102]. Mortality in Chagasic CM is frequently due to sudden cardiac death in 55% to 65% of patients, congestive HF in 25% to 30% of the patients and thromboembolic events in 10% to 15% of the patients but the proportion may vary by the patient population studied [103].
Diagnosis: Diagnosis of Chagasic CM involves laboratory tests for T- cruzi and cardiac investigation both depending on the phase of the disease. Polymerase chain reaction (PCR) is the most sensitive test in acute infection showing rising parasite loads even before they are visible by microscopy [31]. Once parasitemia levels increase, direct microscopy examination of fresh anticoagulated blood or buffy coat can detect motile trypomastigotes [8]. Microhematocrit is a widely used method to identify congenital infection [31]. In the indeterminate and chronic phases, due to low levels of parasitemia, diagnosis is often achieved by using serological tests to detect immunoglobulin G (IgG) that binds T. cruzi antigens. Enzyme-linked immunosorbent assay (ELISA), indirect immunofluorescence (IIF), and indirect hemagglutination (IHA) are the most common methods, and diagnosis is confirmed by two positive tests using any of the three methods [104]. However, cross-reactivity with other parasitic infections and autoimmune diseases may result in poor sensitivity [8,31].
Cardiac investigation aims to detect abnormalities in cardiac function and/or structure. In the acute phase, principal ECG abnormalities include first-degree AV block, QRS and T-wave changes [9]. In the indeterminate phase of infection, ECG or X-ray abnormalities most often are missing, however, stress testing and echocardiography may reveal latent myocardial abnormalities [105]. Echocardiography is thus an important diagnostic modality for the initial assessment as well as long-term follow-up of Chagasic CM patients for predicting mortality and clinical events [106]. LV systolic function is usually preserved in the acute phase unless myocarditis leads to ventricular dysfunction [105]. Advanced Chagas heart disease mimics chronic ischemia or idiopathic DCM. Typical echocardiographic features include apical aneurysm, segmental LV contractile abnormalities, and depressed LV systolic function [91]. Pericardial effusion in acute infection occurs in 42% of patients [8]. Chest X-ray is performed in the posterior-anterior and lateral projections to evaluate atrial and ventricular sizes. Enlargement of all the four cardiac chambers in the absence of pulmonary congestion suggests Chagasic CM [31]. Cardiac magnetic resonance has superior capability for anatomic and functional assessment of all cardiac chambers, provides direct measurement of both RVEF and LVEF, detects mural thrombosis and allows tissue characterization but it has limited availability [31].
Treatment: Management and control of Chagas disease can be categorized into (i) treatment of clinical manifestation, and (ii) control of parasitic infection indirectly through vector control. Antiparasitic drug development is challenging because of the intracellular nature of the parasite. There has been no new drug development since the 1970s [8]. Current antitrypanosomal drug therapies with proven efficacy in the acute phase of Chagas disease are benznidazole and nifurtimox. Benznidazole is the first line of treatment because of better tolerance, widely availability and more data on its efficacy. However, benznidazole has smaller benefits in the chronic phase of the disease. Management of cardiac involvement may include medical, interventional and surgical treatment. The use of beta-blockers it safe and well tolerated. Angiotensin converting enzyme – inhibitors (ACE-I) are additionally used. Amiodarone is used for treatment of arrhythmias and has trypanocidal effects. Cardiac pacing is also often necessary. Implantable cardioverter defibrillator (ICD) is recommended for patients with sustained VT and those successful resuscitated from near sudden cardiac death, consideration for cardiac transplantation for end-stage HF and stem-cell based therapy in very advanced stages of Chagasic CM.
Control of Chagas disease through vector control measures has been successfully in some areas of South America using seroprevalence surveys that seeks to identify areas with the highest risk of infection. High-risk areas may be targeted for vector control measures such as parathyroid insecticidal spraying and improvement of housing conditions – elimination of cracks in mud walls and provision of metal sheet roofing have proved to be effective for reducing the habitat for vectors transmitting Chagas disease [31]. The “Southern Cone Initiative” a vector-controlling programme initiated in the early 1990s, was effective in reducing T. cruzi transmission in many South American countries in which the program was initiated [8]. Vector control strategies used in the program included residual spraying with synthetic pyrethroids, housing improvement and health education. The success of the program was partly attributable to the Triatoma infestans and Rhodnius prolixus (the main transmission vectors) are domiciliated (their life cycles occur completely within domestic habitats) [107]. Universal screening of blood products is also a preventive strategy for transfusion related infections. New housing development, labour-intensive and expensive insecticide spraying regiments and T, cruzi having many animal reservoirs may inhibit the success of vector control programs [108].
African trypanosoma (Human African Trypanosomiasis)
African trypanosoma is a single celled flagellate protozoon responsible for human African trypanosomiasis (HAT; sleeping sickness) in rural populations in sub-Saharan Africa. African trypanosoma species has two forms: (i) Trypanosoma brucei rhodesiense (T. b. rhodesiense) indigenous to East and Southern Africa; and (ii) Trypanosoma brucei gambiense (T. b. gambiense) indigenous to West and Central Africa) and the cause of 97% of HAT cases in Africa [9]. Tanzania and Uganda represent 89% of the total burden of T. b. rhodesiense infection in Africa while the Democratic Republic of Congo, Angola, and Sudan report about 1,000 new cases annually and represent 90% of the total burden of T. b. gambiense infection in Africa [109]. Transmission of the disease is via the bite of Tsetse fly (Glossina species), which acquire their infection from human or from animal hosts that harbour the human pathogenic parasites. The trypanosomes multiply at the site of inoculation – in subcutaneous tissues, blood and lymph – referred to as the haemolymphatic stage characterized by bouts of fever, headaches, joint paints, and itching. Trypanosomes disseminate hematogenously to distant organs resulting in the chronic stage – the meningoencephalitic stage characterized by the invasion of the central nervous system. Congenital and sexual transmission may also occur with African trypanosomes although uncommon [109]. The disease manifests after a variable incubation period in two stages: (i) an early stage involving symptoms of arthralgia, headache, pruritus and lymphadenopathy; and (ii) a late stage predominated with neurological symptoms and high mortality if left untreated. T. b. rhodesiense causes acute or sub-acute syndrome that evolves over days to weeks while T. b. gambiense causes a more protracted clinical course over months to years. Cardiac involvement and manifestations is uncommon in HAT, with studies focusing on signs and symptoms of CNS infection [110]. Thus, the question of whether cases of cardiac involvement in HAT is truly low or the interest on CNS involvement overshadows that of the heart remains unanswered.
Pathophysiology: Neurological problems dominate in HAT while cardiac involvement is uncommon. Animal models and autopsy studies investigating immunopathogenesis of the early and late stages of HAT have made little reference to cardiac pathology effectively undermining the understanding of the pathophysiology of HAT with cardiac involvement. Nevertheless, few and scattered data about pathological cardiovascular features after trypanosomes infection demonstrate cardiac involvement in HAT. Pancarditis has been demonstrated in HAT in experimental model of animals infected with T. brucei [111,112]. Dog and mice experimental models of T. brucei infection suggest lymphocytes and plasma cells infiltration of different cardiac cells followed by the infiltration of macrophages and polymorphonuclear cells in more advanced stages of the disease [111-113]. These cellular infiltrates predominate the endocardium or sub-epicardial and perivascular sites associated with increasing numbers of trypanosomes in the interstitium. Myocardial involvement with degeneration, necrosis, separation and oedema of muscle fibres is also prominent. Trypanosomes and cellular infiltration affect all four cardiac values, the conduction system and lymphatic drainage system of the heart. In the pericardium, blood fluid containing numerous trypanosomes and fibrin have been described as well as microscopic evidence of biventricular dilatation and pericardia fat oedema in advanced stages of the disease [111-113]. Autopsy studies also reveal chronic pancarditis with lympho-mononuclear cellular infiltrates in 67% to 80% of autopsies that assessed for cardiac involvement, which was severe in 10% to 20% of these cases and the probable cause of death [114,115]. Myocarditis, epicarditis and degenerative changes accompanied with cellular infiltration in the cardiac conduction system have been also observed [115]. Microscopic appearance of heart of 14 patients who died of HAT in Uganda showed normal hearts except for valvular fibrosis [115] but hearts of fatal cases of HAT due to T. b. rhodesiense reveal an increase in pericardial fluid and cardiomegaly [116].
Clinical presentation: Patients with chronic infection of T. b. rhodesiense with proven myocardial involvement may show signs of heart failure. During late stages of HAT, a small proportion of these patients may present with signs of cardiomegaly and volume overload such as crackles and peripheral oedema [117]. A prospective study comparing HAT patients due to T. b. gambiense with healthy controls reveal HAT patients exhibit exertional dyspnoea, cough, palpitations, abnormal cardiac rhythms, heart murmurs, hepatojugular reflux, hepatomegaly and peripheral oedema but which resolved with treatment for HAT in the absence of specific HF treatment [118]. Patients with HAT may exhibit significantly elevated serum levels of cardiac biomarkers (N-terminal prohormone brain natriuretic peptide [NT-pro-BNP]) but did not correlate with signs and symptoms of HF except for cardiomegaly [118]. HAT patients in the late stage are more likely to present with ECG abnormalities – low voltage, P-R segment depression and repolarization changes. These ECG abnormalities however do not correlate with laboratory biomarkers of cardiomyocyte necrosis such as elevated troponin levels. However, significant conduction abnormalities are not prevalent findings for HAT patients despite histological evidence of the involvement of cardiac conduction system in both animal models and human autopsy studies [112,118,119].
Diagnosis: Clinical evaluation for CM due to HAT requires demonstrable evidence of T. brucei infection accompanied by evidence of cardiac dysfunction. Diagnosis rely on laboratory examination to detect the parasite in body fluid or tissue by microscopy because the clinical features of the disease are not sufficiently specific [120]. T. b. rhodesiense infection has a significantly higher parasite load than T. b. gambiense infection. AS a result, T. b. rhodesiense is easily detectable in blood as well as in lymph node or in fluid or biopsy of a chancre. Classical diagnosis of T. b. rhodesiense infection is by microscopic examination of the lymph node aspirate usually from a posterior cervical node. However, it is often difficult to detect the parasite in blood thus may require concentration techniques such as capillary tube centrifugation, miniature anion-exchange centrifugation, and quantitative buffy coat smears can be used to improve the sensitivity of the detection of the parasite in the blood and cerebrospinal fluid (CSF) [121-123]. Serological testing such as card agglutination trypanosomiasis test (CATT) is normally preferred for screening purposes while definitive diagnosis rests on microscopic observation of the parasite [120]. Determination of the stage of the disease is important for treatment purposes. Demonstration of CNS involvement rests on detection of the presence of CSF white blood cells count > 5 X 106 cells/litre, CSF protein level > 400 mg/litre or the presence of the parasite [123].
Establishing diagnosis of cardiac involvement in HAT is not straightforward. Few clinical signs and symptoms exist in relation to cardiac involvement. Even when the patient is symptomatic, they are non-specific and establishing their precise aetiology is complicated due to lack of specificity and multiple confounding comorbidities such as anaemia and possible the treatment-induced cardiotoxicity [9]. ECG abnormalities may be common in late stage HAT but it is unclear whether these abnormalities manifest in the early stages of the disease [119,124,125]. HAT CM can be suspected in the presence of parasite and ECG abnormalities including low QRS voltage, PR interval depression and repolarization changes, and evidence of cardiomegaly on imaging or on physical examination. Confirmatory diagnosis would however require histological evidence of cardiac inflammation although endomyocardial biopsy is not advisable because the risks are likely to outweigh the benefits [9]. Treatment: Treatment for HAT CM takes two forms: (i) aetiological; and (ii) symptomatic. Aetiological treatment targets the elimination of the causative parasite while symptomatic treatment targets symptoms of cardiac dysfunction as well as of infection. Infection with T. brucei if untreated has almost a 100% fatality [126]. Usually, treatment varies based on the aetiological agent and the stage of the disease. Treatment recommendations for the early stage of the disease is suramin for T. b. rhodesiense and pentamidine or suramin (an alternative) for T. b. gambiense while in the late stage (CNS infection) is melarsoprol (for both parasites) or eflornithine (only for T. b. gambiense). Both melarsoprol and eflornithine are potentially cardiotoxic, and newer treatment alternative such as a dual therapy of the melarsoprol and eflornithine or nifurtimox is ongoing [9]. Symptomatic treatment of cardiac dysfunction is usually not necessary. Signs and symptoms of HF are relatively mild, have no correlation with ECG abnormalities or laboratory markers of LV dysfunction, and aetiological treatment can cause ECG abnormalities. More importantly, non-specific signs and symptoms of LV dysfunction often resolve with aetiological treatment of HAT [118]. Finally, prophylactic measures in countries where HAT is endemic focus on systematic population screening to identify and institute treatment to reduce human reservoir and vector control. Other preventive measures include avoiding travel in endemic areas or wearing heavy fabrics with neutral colours covering as much skin as possible [127].
Toxoplasma (Toxoplasmosis)
Toxoplasma gondii (T. gondii) is an obligate intracellular parasitic one-celled eukaryote with a worldwide distribution. It is the aetiologic agent of the infectious disease toxoplasmosis. Felines are the definitive hosts with the domestic cat being the most important. T. gondii occurs in mature in three forms: (i) oocysts found only in felines and eliminated in their excreta and represents the source of infection for susceptible hosts; (ii) tachyzoites/intracellular proliferative form present during the acute phase of infection ; and (iii) tissue cysts responsible for the latent infection of multiple organs and important for disease transmission [128]. Pathways of human infection are the ingestion of cysts in raw or uncooked meat, mainly pork and beef; ingestion of mature occysts in food or water contaminated with cat excreta or via vertical transmission through transplacental passage of the parasite from mother to foetus with acute infection of the foetus [129]. Although less common, transmission can occur via blood transfusion, laboratory accidents and organ transplantation [130].
gondii can infect and cause clinically distinct syndromes in both immunocompetent and immunocompromised individuals. In immunocompetent hosts, the infection is frequently benign with self-limiting parasitemia, which in most cases results in asymptomatic clinical form of toxoplasmosis. However, in about 20% of the cases, acute infection may manifests with febrile lymphadenopathy, asthenia and lymphomonocytosis, with a self-limiting course of infection [131]. After the acute infection phase, T. gondii remains viable in the form of tissue cysts, which reproduce slowly throughout the life of the host, and thus, characterized the chronic phase of injection. In the chronic phase, humoral and cellular immune system (T lymphocytes and macrophages) stimulated by parasites antigens, controls tissue cysts and re-infection. In less immunologically active tissues such as the CNS, parasite multiplication may be more active and persists for longer periods [132]. However, immunocompromised hosts are at risk of recurrence of chronic infection and dissemination, with the occurrence of fulminating disease. T. gondii most frequently infects the CNS and less frequently the heart and lungs [133-135].
Pathophysiology: T. gondii multiplies intracellularly at the site of inoculation and disseminate to distant organs via the blood and lymphatics. Tissues cysts form within the first week of infection and are responsible for latent infection. The parasite lives intracellularly in phagosomes within macrophages and myocardial cells. T. gondii is propelled by an actin-myosin-dependent gliding motility mechanism to establish intracellular vacuoles. This remodelling prevents lysosome fusion leading to intracellular survival of the parasite [99]. The immune competency of the host is a key factor that determines the recurrence of latent infection since immunity in the immunocompetent host persists for life. T lymphocytes, macrophages and type 1 cytokines are critical for controlling infection by T. gondii. Defective production of interferon gamma (IFN-γ) and interleukin-12 may reduce the effectiveness of immune response to control T. gondii infection [136,137]. Host genetics may play a role in the disease pathogenesis. HLA-DQ3 has been associated with the development of toxoplasmic encephalitis and the possible protective role of HLA-DQ1 to suggest host genetic factors may contribute to the development of the disease [138-140].
Clinical presentation: Cellular immunity is the principal mechanism of defence in the control of toxoplasmosis. Thus, clinical presentation of toxoplasmic CM may vary considerably depending on the levels of immune competency of the infected host. Acute toxoplasmosis is often asymptomatic in immunocompetent hosts but with lifelong latent infection. In contrast, in immunocompromised patients, latent infection due to cyst formation may reactivate or may progress into encephalitis or chorioretinitis [141-144]. Some patients may present with myocarditis, pericardial effusion, constrictive pericarditis, arrhythmias and HF [142,145]. In AIDS patients, the heart is the second most frequently infected organ after the brain [141,142]. However, confirmation of diagnosis is made at autopsy because signs and symptoms of cardiac involvement are usually silent or may be dominated by CNS manifestations [100]. Besides AIDS patients, other immunocompromised patients that toxoplasmosis may be suspected are transplant patients due to reactivation or de novo infection from a seropositive donor to seronegative recipient [146,147]. Toxoplasmosis is prevalent in heart transplant patients and may stimulate organ rejection and disseminated toxoplasmosis with myocarditis without treatment may be fatal [17,144,148].
Diagnosis: Clinical evaluation and detection of toxoplasmic CM rests on a combination of clinical and laboratory data. The basis of diagnosis is the evidence of tachyzoites in myocardial tissue and cardiac dysfunction. In clinical practice, routine use of serological tests are routinely is able to detect IgM and IgG specific antibodies including immunofluorescence and immune-enzymatic tests (ELISA), with the latter showing high sensitivity and specificity [132]. A negative IgG antibody test on immunocompetent host allows the exclusion of prior or recent infection because IgG antibodies appear early after infection, peak within six months and remain detectable for life [149]. On the other hand, IgM antibodies persist years after infection and not advisable as the sole diagnostic test for recent infection [149]. Antibody avidity (the strength with which an antibody binds to a complex antigen) is proving a useful market of the timing of infection. High avidity indicates a recent infection that occurred 3 to 5 months. However, no single serological test supports a definitive diagnosis of acute or chronic toxoplasmosis, and the use of reference laboratory data is often required.
Some patients may exhibit laboratory findings such as slight lymphocytosis and slightly elevated hepatic transaminase levels as well as fluctuating ST changes on ECG, but all are non-specific to support diagnosis [150,151]. Endomyocardial biopsy (EMB) has been successful in detecting infective organisms in heart transplant patients [144,152,153] and the benefits of early and specific EMB diagnosis in heart transplantation may outweigh the potential risks. Typical risks of EMB include local necrosis with oedema and inflammatory infiltrate [154]. Although myocardial abscesses have been reported, they are uncommon pathological feature of toxoplasmosis [100,154]. Other less common tests to detect the presence of T. gondii include DNA implication techniques and PCR, which has been shown to enable the diagnosis of toxoplasma more frequently relative to cardiac biopsy specimen in heart transplant settings [155]. Thus, PCR assays can be valuable in the diagnosis of toxoplasma and encourages the use of reference laboratories with experience in these assays.
Treatment: Clinical management strategies for toxoplasmic CM include treating T. gondii infection and prevention against infection. Medical therapies of choice for toxoplasmosis is a dual therapy of pyrimethamine and sulfadiazine (first-line treatment in pregnant women), or pyrimethamine and clindamycin [148,156]. Folinic acid (Leucovorin) reduces the toxic effects of pyrimethamine and should be given to all patients on pyrimethamine. For intolerant patients, a dual therapy of pyrimethamine and azithromycin or atovaquone may be considered [9]. Preventive strategies include serological screening of donors and recipients before transplantation to identify patients at risk of toxoplasmosis such as seropositive recipients and mismatched solid-organ recipients (seropositive donor and seronegative recipient). Prevention in transplant patients rests on prophylaxis using cotrimoxazole [157]. Preventive strategies may also include patient education on the avoidance of materials potentially contaminated with feline excreta, use of gloves when handling cat litter or gardening, washing hands thoroughly after handling raw meat, proper cooking of meat and washing fruits and vegetables before consumption [157].
Plasmodium (Malaria)
Plasmodium is a single-celled eukaryotic protozoan parasite that multiples in red blood cells of humans as well as in mosquito intestines. Plasmodium that infects humans cannot infect animals and thus, humans are the only known reservoir. Four different types of Plasmodium cause malaria in humans under natural conditions: P. falciparum, P. vivax, P. ovale, and P. malariae. However, only the P. falciparum and P. vivax can cause severe malaria, which can result in death if adequate treatment is not provided promptly. P. ovale and P. malariae have a dormant phase of their lifecycle and can be present in human host for months to years with no symptoms [158,159]. The principal pathway of Plasmodium transmission in humans is the bite of the female Anopheles mosquito [159]. P. falciparum is the only infection that can cause severe malaria with cardiac involvement [160-162].
Pathophysiology: Impaired pre- or post-cardiac circulatory parameters or myocardial dysfunction itself has been implicated as underlying pathological mechanisms for cardiac dysfunction in malaria patients. Intravascular fluid depletion may lead to impaired microcirculation, reduced preload and consequently low cardiac output. Other factors contributing to rapid fluid loss are high body surface (in children), high fever, diarrhoea, vomiting and limited intake of fluids [158]. Significantly increased peripheral vascular resistance may also contribute to low cardiac output. Typical pathophysiological mechanisms of P. falciparum is parasite adhesion to the endothelium, resetting, sequestration of parasitized and unparasitized red blood cells in peripheral small vessels and reduced deformability of red blood cells leading to impaired microcirculation and lactic acidosis [157,158,160]. NT-proBNP is significantly elevated in patients with severe malaria suggesting a direct effect of Plasmodium on myocardial function. Plasmodium toxins or host immune mediators, or both exert a suppressive effect on myocardial function. Plasmodial toxin glycosylphosphatidylinositol (GPI) augments apoptosis rates in cardiomyocyte culture. Secondary infections, severe anaemia, hyperpyrexia, dehydration or fluid overload, metabolic acidosis, hypoxia and disseminated intravascular coagulation may exacerbate cardiac dysfunction in malaria [160]. The host immune reaction targeting malaria parasites involves pro- and anti-inflammatory cytokines and immune mediators such as nitric oxide (NO) [160]. Pro-inflammatory cytokines and immune suppress myocardial function while cardiac biomarkers are good prognostic markers for outcomes in septic and critically ill patients. Anti-malarial drug may also exert additive cardiotoxic effects [158].
Diagnosis: The gold standard test for diagnosing of Plasmodium parasite is blood smear usually stained with Giemsa stain and observed under 100X oil immersion. The early trophozite form of the Plasmodium can be observed in red blood cells and has a characteristic ring shape. P. falciparum is distinguishable by more than one visible in the red blood cell while the other three species there is only one visible [163]. There is evidence of manifestations of serious symptoms of coronary complications in Plasmodium infection [164,165]. ECG abnormalities have been described in patients with severe malaria including deaths due to cardiac arrhythmias [166-171]. Serum levels of cardiac biomarkers such as troponin T, myoglobin and creatine kinase increase with severity of malaria indicating myocardial dysfunction in P. falciparum malaria [172,173]. Although cardiac troponin is a sensitive biomarker for the detection of minimal myocardial damage due to a variety of conditions including inflammation, trauma, exposure to toxins and necrosis because of occlusion of a coronary vessel [174,175], elevated troponin levels is rare in patients with P. falciparum malaria [173]. Evidence of upregulated gene expression of cardiomyocytes related to apoptosis and myocardial damage treated with purified P. falciparum glycosyl phosphatidylinositol (a toxin in malaria pathogenesis) suggests P. falciparum can induce apoptosis [176]. Case reports and rodent models also describe acute coronary syndrome, tachycardia, arrhythmias and myocardial failure in malaria patients [177-180].
Treatment: Clinical management of Plasmodium CM rests on treatment and elimination of the causative parasite. Upon the confirmation of diagnosis, the appropriate antimalarial treatment must be initiated immediately. Three key clinical factors should guide treatment: (i) the infecting species of Plasmodium; (ii) the clinical condition of the patient; and (iii) the susceptibility of the parasite to the anti-malarial drugs determined by the geographical area where the infection was acquired and previous use of the medicine [163,157]. Most anti-malarial medication such as chloroquine, mefloquine, quinine, quinidine, doxycycline, clindamycin and artemisinins act against the early stages of infection that causes the symptoms [157].
Leishmania (Leishmaniasis)
Protozoans of the genus Leishmania are a diverse group of intracellular parasites transmitted to mammalian hosts by infected blood-sucking sandfiles (Phlebotomus species). Various animals serve as reservoir host of Leishmania including humans, domestic and feral dogs, rodents, foxes, jackals, wolves, raccoons and hyraxes [181]. Less common non-vector transmission pathways include blood transfusion, sexual intercourse, organ transplants, excrements of dogs, and sporadically outside endemic areas [182]. Human infection by pathogenic Leishmania result in Leishmaniasis, a disease describing cutaneous and visceral infections [181,182]. The estimated global annual incidence of Leishmaniasis is 2 million across 98 countries with an additional 350 million at risk of infection [183]. Leishmania have two main lifecycle stages: (i) the motile flagellated promastigote present in the sandfly vector; and (ii) intracellular non-flagellated amastigote present in the mammalian host cells.
Pathophysiology
Cardiac involvement in Leishmania infection is uncommon and its pathophysiology not well defined. Generally, Leishmania are parasites of phagocytes (macrophages and dendritic cells). They initiate infection via receptor-mediated binding of infective promastigotes delivered into the host during feeding of infected sandfiles. Parasites contained in parasitophorous vacuoles fuse with lysosomes to form phagolysosome wherein promastigotes transform into and replicate as amastigotes [184]. Eventually the parasite burden increases physically disrupts macrophages of the infected host to deliver extracellular amastigotes into the surrounding tissues, where uninfected macrophages destroys them. Leishmania and infected macrophages can metastasize within the skin and visceral organs [185,186]. The effect of Leishmania as well as host immune system in myocardial function has not been described in humans. However, in a canine model of Leishmaniasis, an important histological finding was the presence of intense and chronic inflammatory reaction composed of mononuclear cells (predominantly monomorphic macrophages, as well as plasma cells and lymphocytes) in most organs. In the heart, the myocardium has a dense accumulation of macrophages between muscle fibres and areas of cardiac muscle atrophy, degeneration and loss of cardiomyocytes [187]. Two cases of cardiac involvement in Leishmaniasis have been reported in India and Iran involving heart failure (Iran) and heart failure and pericardial effusion (India) [188].
Diagnosis
Clinical evaluation for Leishmaniasis is difficult because of the various forms of the disease, variety heterogeneity of parasites involved, geographic variations and other clinically similar syndromes such as malaria, typhoid fever, typhus, and schistosomiasis. Confirmatory diagnosis of Leishmaniasis rests on microscopic identification of Leishmania in liver, spleen or bone marrow or by the detection of DNA of Leishmania by PCR in blood or biopsy material [182]. Reported echocardiographic evidence of cardiac dysfunction include pericardial effusion in a patient living in Leishmaniasis endemic area [182]. In a study of echocardiographic evaluation of cardiac status in 14 Indian visceral Leishmaniasis patients, LV function and dimensions remained within normal limits in all patients but pericardial effusion was observed in four patients with heavy parasitemia. Effusions were small, haemodynamically insignificant and resolved spontaneously [188].
Treatment
Clinical management of Leishmaniasis targets elimination of the causative parasite since associated cardiac dysfunction often resolved spontaneously [188]. Treatment may range from local treatment of cutaneous lesions to systemic therapy for disseminated cutaneous and deadly visceral disease. A number of therapies are available and preference for first-line and second-line treatment may vary based on the type of disease and often guided by regional practice [181]. Pentavalent antimony is considered the mainstay of Leishmaniasis therapy for decades although it has been associated with multiple toxicities and is increasingly ineffective due to growing parasite tolerance. Other alternative medications used in different clinical situations and guided by availability and effectiveness in different localities include Amphotericin B, Paromomycin, Pentamidine, Miltefosine, Imiquimod, Azoles, or Cryotherapy [181].
Other Protozoan Parasites
Protozoan parasitism covers a broad spectrum of diseases but Entamoeba, Balantidium and Sarcocystis barely register among infectious protozoan diseases particularly in myocardial involvement. However, their consideration as protozoan aetiologies of cardiomyopathy is important to aid in diagnosis and clinical management in endemic areas.
Entamoeba (Amebiasis)
Entamoeba histolytica is a pathogenic protozoan transmitted via the faecal-oral route usually residing in the large bowel. It has a worldwide distribution but more frequent in developing countries and in the tropics [189,190], where it is the third most important cause of parasitic death after malaria and schistosomiasis [191]. High-risk groups include children pregnant women and malnourished individuals [9]. Cardiac involvement manifesting as pericarditis is rare but a serious complication of liver abscess [192]. E. histolytica is morphologically identical to E. dispar but differs because of its ability to invade tissues [193]. In most of the infected patients, E. histolytica causes acute inflammation and ulceration of the colonic mucosa or rectocolitis [10]. Adherence lectin (Gal/GalNAc: a virulent factor of E. histolytica as well as essential for its pathogenesis) mediates colonization to colonic mucosal cells and mucosal IgA immunity to this lectin protects from Entamoeba infection and disease [194,195]. The trophozite uses the secretion for proteinases to dissolve the extracellular matrix to form amebepores and cause target cell cytolysis to establish its pathogenetic niche [196-198].
Cardiac involvement in amebiasis is rare but usually affects the pericardium presenting as pericardial run with ECG changes associated with an abscess of the left lobe or purulent pericarditis from the perforation of the abscess into the pericardium [199-200]. Clinical presentation may be a sudden onset of cardiac tamponade with chest pains, dyspnoea and shock, or progressive effusion with a slower course to develop fever, dyspnoea and pain [201,202]. Diagnosis of amebiasis rests on serology. E. histolytica leads to the development of antibodies while E. dispar does not. However, positive serology does not distinguish between acute and past infection and thus serology is used to support diagnosis. Stool microscopy has limited value for diagnosing extra-intestinal amebiasis because amoebic cysts or trophozite are detected in only 15% to 33% of the time [203]. Due to the limitation of serology tests, PCR-based diagnostic methods are becoming invaluable sensitive and specific for detecting intestinal and extra-intestinal amebiasis [204,205]. Chest computed tomography and echocardiography might be useful to detect liver abscess in continuity with the pericardium and fluid within the pericardial sac [206]. Clinical management of amebiasis with cardiac involvement rests on a combination of surgical drainage and metronidazole. It clinical improvement does not occur within 48 to 72 hours, bacterial superinfection should be considered, and treated if found, and metronidazole therapy prolonged [207].
Balantidium (Balantidiosis)
Balantidium Coli is the only and the largest parasitic protozoan of the ciliate phylum with a worldwide distribution known to be pathogenic to humans. The parasite is usually observed in two stages: the mobile trophozite stage, sensitive to desiccation, and the cyst stage, that can remain viable up to two weeks in the environment. Although rare in high resource countries, balantidiosis is a zoonosis disease acquired by infection of Balantidium to humans via the faecal-oral route from its normal reservoir, the pig, where is it asymptomatic. Pigs pass Balantidium in their excreta, which can contaminate wells and ground water, serving as the principal vehicle for transmission of the parasite [208]. In humans, Balantidium inhabits the cecum and the colon, where is causes asymptomatic infections or may develop dysentery similar to that caused by E. histolytica. Rarely, Balantidium invades extra-intestinal sites such as the liver, lung, tract, heart and causes infection [209,201]. Other intestinal infection or parasites, malnutrition, alcoholism, immunodeficiency or a history of chronic disabling disease render patients more vulnerable to Balantidiosis. Cardiac infection is rare but has been reported with moderate serious atrophy of the sub-epicardial fat and a slight diffuse infiltration of the myocardium with lymphocytes and eosinophils, as well as slight interstitial oedema [209,211]. In a majority of cases, Balantidiosis is asymptomatic. Clinical manifestations include persistent diarrhoea, occasionally dysentery, abdominal pain, and weight loss. Clinical evaluation of Balantidiosis rests in detection of trophozoites in stool specimens or in tissue collected during endoscopy. Cysts are less frequent since humans pass Balantidium Coli intermittently and once outside the colon is rapidly destroyed. Thus, stools specimen should be collected repeatedly and immediately examined or preserved to enhance detection of parasite. Common medications used for Balantidium Coli include tetracycline, metronidazole, and iodoquinol [208, 210].
Sarcocystis (Sarcocystosis)
Sarcocystis species are intracellular zoonotic protozoan parasites of the phylum Apicomplexa. Sarcocystis requires two separate hosts a definitive host (in which the sexual stage develops usually a carnivorous predator) and an intermediate host (often an herbivorous prey) to complete its life cycle. The definitive-host infection is limited to the gastrointestinal tract (intestinal sarcocystosis), whereas intermediate-host infection leads to the formation of characteristic intramyocytic cysts (sarcocysts). Although Sarcosporidiosis was once considered rare in humans, outbreaks of symptomatic intermediate-host disease (muscular sarcocystosis) among tourist in Malaysia suggest Sarcocystis infection is an increasing public health problem. The definitive host (often a carnivore) becomes infected by eating tissue from an intermediate host that contains sarcocysts whereas the intermediate host is infected by consuming contaminated water. Once ingested, the sporoziotes penetrate the mucosa of the small intestines and lodge in the vascular endothelial cells. Damaged to the vasculatures results in haemorrhage and anaemia. The parasite ultimately enter the muscle and nerve cells, where they develop into sarcocysts [212,213]. Definitive diagnosis requires detection of sporocysts in faeces using several stool examinations beginning several days after eating raw or undercooked meat. Diagnosis of Sarcocystis in muscle biopsy specimens requires microscopic examination of histologic sections stained with haematoxylin and eosin [212]. There is no known prophylaxis or therapy for intestinal sarcocystosis since infection are often self-limiting, short duration and often asymptomatic. While medication such as co-trimoxazole or furazolidone could be prescribed, their efficacy has not been demonstrated [212].
Meta-analysis of diagnosis/management
Infectious cardiomyopathy remains a significant cause of HF, particularly in endemic areas or in resource-constrained countries. Its management is complicated by a wide heterogeneity of infectious aetiologies. However, the bulk of the current published evidence has centred on viral and bacterial aetiologies, which has undermined the exact understanding of other less common but potentially deadly pathogenic micro-organisms including protozoa parasites. Thus, the present meta-analysis pools published evidence on diagnosis and management of protozoal CM. The search for relevant studies was performed on online database PubMed from inception to April 2019. Further, a manual search of references of the included studies and review articles was to identify additional studies that may have been overlooked by the initial electronic search. The inclusion criteria were studies that included patients (i) diagnosed with protozoan infection with cardiac involvement; (ii) using serological and PCR tests, ECG or non-invasive imaging (ECG or echocardiography); or treated using medical therapy for the infectious agent or heart failure. Studies using animal models, case reports, conference papers and review articles were excluded. In total, the present meta-analysis included 27 studies published between 1985 and 2017 [161,170,171,174,214-236]. Table 3 provides a summary of the key characteristics of the study design, study population, diagnosis tests and findings.
Table 3. Summary of the included studies
First author (Year) [Ref #] |
Year |
Study design |
Primary infection |
No. of patients |
Diagnostic tests |
Summary of main findings |
Sosa-Jurado [214] |
2003 |
Cross-sectional |
T. cruzi |
45 |
Serological, ECG |
20-22% of seropositive individuals have ECG abnormalities (RBBB or ventricular systoles) more than seronegative individuals (p<0.05). Serologic status influences frequency/type of ECG abnormality but not endemicity |
Angel [215] |
2004 |
Cross-sectional |
T. cruzi |
486 |
Serological, ECG |
Seropositive individuals have a higher incidence of RBBB, AV block, ventricular extrasystoles, inverted T-wave and bordering PR (p< 0.001) than seronegative |
Goldbaum [216] |
2004 |
Cross-sectional |
T. cruzi |
365 |
Serological, ECG |
A higher proportion of seropositive workers (42.7%) have ECG abnormalities compared to seronegative workers (19.8%) |
Becerril-Flores [217] |
2007 |
Cross-sectional |
T. cruzi |
8 |
Serological, ECG |
Virulence of T. cruzi is associated with variation in the risk of infection and human seroprevalence that ranged between 3.25 and 5.13% |
Williams-Blangero [218] |
2007 |
Cross-sectional |
T. cruzi |
722 |
Serological, ECG |
Seropositive persons have significantly longer QRS (98 vs. 91 ms) and QT intervals (397 vs. 382 ms), and more frequently conduction abnormalities (43% vs. 18%) than seronegative persons |
Borges-Pereira (2008) [219] |
2008 |
Cross-sectional |
T. cruzi
|
17 |
Serological, ECG |
Chagas has a seroprevalence of 3.1% higher among adults > 50 years old. In seropositive patients parasitemia occurred in 11.8% by indirect xenodiagnoses; 75% by PCR; and in cardiopathy in 41% by anamnesis, physical examination and resting ECG. |
Brum-Soares [220] |
2010 |
Prospective |
T. cruzi |
38 |
Serological, ECG, Echo |
More seropositive individuals had ECG abnormalities (37% vs. 22%) and echocardiography (32% vs. 18%) compared to seronegative individuals |
Moretti [221] |
2010 |
Cross-sectional |
T. cruzi |
425 |
Serological, ECG |
Seroprevalence higher in rural areas (65-89%) than urban (57%). ECG abnormalities higher in seropositive (27%/33% vs. 17/22%) for seronegative patients |
Silva [222] |
2010 |
Prospective |
T. cruzi |
14 |
Serological, ECG |
ECG abnormalities occurred frequently, and conduction disorders of the right branch predominated. |
Tobar [223] |
2011 |
Cross-sectional |
T. cruzi |
135 |
Serological, ECG |
Seroprevalence for antibodies for T. cruzi higher in HF patients (40%) than healthy control (11%) and prolonged QRS, decreased LVEF, high serum magnesium |
Monteon [224] |
2013 |
Retrospective |
T. cruzi |
128 |
Serological, ECG, PCR |
Triatoma dimidiata infected with T. cruzi I and feed on human beings with relative high frequency, but seroprevalence and Chagas disease in humans is relatively low |
Ribeiro [225] |
2013 |
Retrospective |
T. cruzi |
499 |
Serological, ECG |
RBBB and LAFB are significantly common in seropositive individuals (p<0.0001) as well as rhythm disorders, intraventricular blocks and ischemic abnormalities |
Molina-Garza [226] |
2014 |
Cross-sectional |
T. cruzi |
2688 |
Serological, ECG (52 were positive) |
Seropositive rate 1.93% with 23% of infected individuals showing ECG abnormalities. Risk factors: ceiling construction material (p ≤ 0.0024), domestic animals (p ≤ 0.0001), and living in rural municipalities (p ≤ 0.0025) |
Ribeiro [227] |
2014 |
Prospective |
T. cruzi |
557 |
Serological, ECG |
Patients with Chagas disease has more frequent ECG abnormalities (88%) than non-infected (78%) p<0.001; RBBB with left anterior hemiblock is associated with Chagas disease (R: 11.99; 5.6 to 25.7); ECG abnormalities doubles the risk of dearth (HR: 2.18; 1.35 to 3.53) |
Alroy [228] |
2015 |
Cross-sectional |
T. cruzi |
90 |
Serological, ECG |
The seroprevalence of T. cruzi among is 14.9% (95% CI: 12.2 – 18.0%). Independent correlates of infection are increasing age, positive traitomine in a participant's house, and ownership of a T. cruzi positive guinea |
Fernandez [229] |
2015 |
Prospective |
T. cruzi |
753 |
Serological, ECG + Echo (398) |
Overall 13.8% had ECG abnormalities: BBB (11.3%); rhythm disturbances of ventricular ectopy (3.3%); EV blocks (2.6%): ECG abnormality increases with age: 1.1% (10-19 years) to 26.4% (> 60 years) |
Yager [230] |
2015 |
Cross-sectional |
T. cruzi |
604 |
Serological, ECG + Echo 183/421 |
Seroprevalence is for T. cruzi 30% and these patients were more likely to have conduction system defects: compete RBBB (1.6%) and 10.4% for any bundle branch |
Combellas [231] |
1985 |
Prospective |
T. cruzi |
20 |
Echo |
Early isovolumic relaxation and left ventricular abnormalities were pronounced in the patients with Chagas's heart disease and may precede systolic compromise, which may become apparent in later stages of the disease |
Barros [232] |
2004 |
Prospective |
T. cruzi |
169 |
Echo |
21.3% has diastolic dysfunction, associated with worsening LVEF. TDI parameters are useful in detecting diastolic dysfunction with high sensitivity, specificity and negative predictive value |
Viotti [233] |
2004 |
Prospective |
T. cruzi |
849 |
Echo |
LV systolic dimension (3.06 cm) and dysfunction (0.17) predictors of mortality; 0.8% had normal ECG |
Rassi [234] |
2014 |
Retrospective |
T. cruzi |
60 |
Echo |
LVEF 26.6±5.34 % index left atrial volume an independent predictor of death. |
Sánchez-Montalvá [235] |
2016 |
Prospective |
T. cruzi |
485 |
Echo |
31.5% had at least one ECG abnormality and 5.3% with abnormal echo. Patients with abnormal echo were older (47 vs. 41 years) and more likely male (67 vs. 30%) compared to healthy controls |
Gunther [171] |
2003 |
Retrospective |
P. Falciparum |
161 |
ECG/Cardiac biomarkers |
Troponin T was elevated in 0.6%, CK-MB in 0% and myoglobin in 6.2% of the patients. ECG abnormalities found in 14.3% of the patients |
Franzen [236] |
1992 |
Prospective |
P. Falciparum |
22 |
ECG, Echo |
ECG abnormality found in 22.7%; pericardial effusion in 9.1%; and global LV hypokinesia in 4.5%. both ECG and echo normalized in all patients after 19-months follow-up |
Lang [174] |
2000 |
Prospective |
P. Falciparum |
24 |
Cardiac biomarkers |
CK and CK-MB mass and myoglobin assays significantly increased at 10 minutes after biopsy but remained within reference range. Bedside Troponin T indicate myocardial injury in 50%. |
Nayak [161] |
2013 |
Prospective |
P. Falciparum |
17 |
Blood smear, PCR, ECG + Cardiac biomarkers |
Cardiovascular involvement occurred in 17% of cases: circulatory failure 11%, congestive HF 7% and pulmonary oedema 2%; ECG-tachycardia in 17 patients; Troponin and CK-MP in 14 cases. |
Ray [170] |
2017 |
Prospective |
P. Falciparum |
27 |
ECG + Echo |
ECG sinus bradycardia (7%); tachycardia (3.7%); premature arterial ectopic (3.7%). Echocardiographic – global hypokinesia with decreased LVEF (11.1%) LV diastolic dysfunction (3.7%); mild tricuspid regurgitation (3.7%) and mild pericardial effusion (3.7%). |
CK: Creatine Kinase; ECG: Electrocardiography; Echo: Echocardiography; HF: Heart Failure; LV: Left Ventricular; LVEF: Left Ventricular Ejection Fraction; PCR: Polymerase Chain Reaction; RBBB: Right Bundle Block Branch; TDI: Tissue Doppler Imaging
The 27 studies included in this meta-analysis adopted various study designs. The majority were prospective cohort (n = 12; 44%) [161,170,174,220,222,227,229,231-233,235,236] followed by cross-sectional studies (n = 11; 41%) [214-219,221,223,226,228,230] and retrospective cohort (n = 4; 14%) [171,224,225,234]. The greater majority (n = 22; 77%) investigated Chagasic CM due to T. cruzi infection [214-235] and the remaining 23% investigated malaria CM due to Plasmodium infection [161,170,171,174,236]. The common diagnostic tests across all the 27 studies were ECG to evaluate electrical cardiac function, serological to identify the causative protozoan, cardiac biomarkers to evaluate cardiac dysfunction and echocardiographic cardiac imaging to detect alterations in cardiac function and morphology. Altogether, the 27 studies enrolled 9,408 individuals diagnosed with protozoan infection of the heart. In sixteen (16) of the 27 studies [161,170,171,174,218,223,225-231,233-235], both genders had an almost equal representation with male accounting for slightly less than half (48%) of the enrolled patients. The patients were relatively young (48 years; range = 16-68).
Study findings
The choice test for identifying the underlying causative pathogenic protozoa in the included studies were serological tests, most frequently ELISA, indirect hemagglutination and/or indirect immunofluorescence or blood smear for plasmodium. Serological tests detect immunoglobulin G (IgG) antibodies to the aetiologic protozoan parasite. The tests were useful for selecting and classifying patients into either seropositive or seronegative arm of study for comparison. Pooled analysis of 21 studies [161,171,214-230,235,236] to determine the incidence of ECG abnormalities in seropositive patients revealed 30.4% of the patients (95% CI: 21.1% to 41.7%; p = 0.001) exhibited at least one ECG abnormality (Figure 2). Further pooled analysis of 13 studies [214-223,225,226,228] comparing the incidence of ECG abnormalities between seropositive and seronegative patients revealed seropositive individuals have a 2.6 time more likely to exhibit ECG abnormalities compared to seronegative individuals (Odds Ratio [OR]: 2.57; 95% CI: 1.734 to 3.820; p = 0.000) (Figure 3).
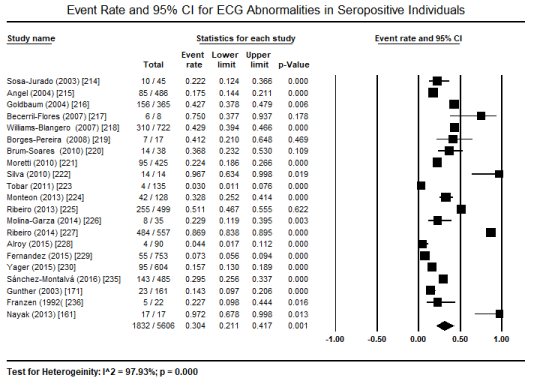
Figure 2. Clinical phases of chagasic cardiomyopathy
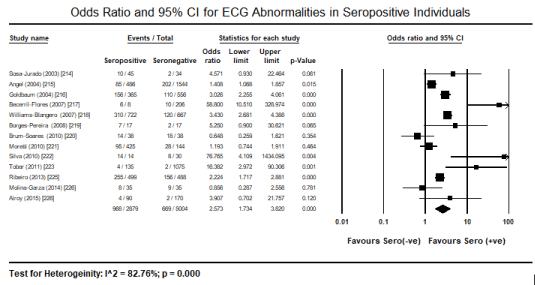
Figure 3. Forest plot for odds ratio for ECG abnormalities in seropositive individuals
Further, pooled analysis of individual ECG abnormalities in seropositive individuals indicate that the most frequently encountered ECG abnormalities are ST-T changes (12.5%) in 3 studies [171,227,235]; RBBB (4.0%) in 7 studies [221,223,225,227,229,230,235]; AV block (2.4%) in 5 studies [223,225,227,229,230]; LV hypertrophy (2.3%) in 6 studies [221,223-225,227,235]; bradycardia (2.2%) in 6 studies [170,221,225,227,229,235]; rhythm disturbance/ectopic and atrial fibrillation/flutter (1.8% each) in 5 studies 5 [170,225,227,229,230]; LBBB (1.3%) in 5 studies [221,223,225,227,235]: low QRS voltage (1.1%) and sinus tachycardia (0.8%) in 4 studies [170,221,225,227] (Table 4).
Table 4. Summary of pooled analysis of the rate of ECG changes in seropositive patients
ECG changes |
No. of studies |
Events/Total |
Event rate (%) |
95% CI (%) |
p-value |
ST-T Changes |
3 |
353/1203 |
12.5 |
1.0 to 67.9 |
0.157 |
RBBB |
7 |
211/3458 |
4.0 |
1.4 – 10.8 |
0.000 |
AV Block |
5 |
92/2548 |
2.4 |
0.8 – 6.9 |
0.000 |
LVH |
6 |
55/2229 |
2.3 |
0.6 – 7.9 |
0.000 |
Bradycardia |
6 |
64/2746 |
2.2 |
1.0 – 4.6 |
0.000 |
RD/Ectopic |
5 |
76/2440 |
1.8 |
0.5 – 6.8 |
0.000 |
AF |
5 |
65/2429 |
1.8 |
0.4 to 6.7 |
0.000 |
LBBB |
5 |
51/2101 |
1.3 |
0.2 – 7.1 |
0.000 |
Low QRS |
4 |
24/2145 |
1.1 |
0.4 – 2.9 |
0.000 |
Tachycardia |
4 |
12/1508 |
0.8 |
0.2 to 3.4 |
0.000 |
Compared to ECG, the present meta-analysis included fewer studies on echocardiographic evaluation of cardiac function and structure in patients with protozoan infection and cardiac. Pooling data on these echocardiography studies was not possible because the presentation was in different formats (number of patients affected or the mean values of LV ejection fraction and LV dimensions. However, echocardiograph abnormalities were common in 5.3% of patients with Chagas disease, mostly males (67%) and older (mean 47 years) in which diastolic dysfunction predicted poor prognosis [235]; significantly delayed aortic valve closure and mitral valve opening during isovolumic relaxation (which is prolonged); increased LV cavity size during isovolumic relaxation and abnormally reduced during contraction; and decreased peak rate of posterior wall thinning and prolonged, which preceded diastolic dysfunction [213]. Diastolic dysfunction is common in 21.3% of patients with a string correlation between worsening diastolic dysfunction and LVEF (r = 0.78). TDI septal e’ is the best methods for detecting any kind of diastolic dysfunction while septal E/e ratio for detecting advanced diastolic dysfunction [232]. Independent predictors of poor prognosis or death include indexed left atrial volume (values > 70.71 mL/m²) are associated with increased mortality [234] LVEF, LV systolic dimensions predict mortality and clinical events [233]. Finally, in asymptomatic patients, treatment focused on eliminating the infectious protozoan and HF therapy in symptomatic patients.
The present systematic review and meta-analysis sought to evaluate common diagnostic and treatment methods of protozoan CM. The search for relevant publications revealed a paucity of studies on protozoan CM suggesting it remains a grossly under-researched aetiology of CM relative to viral and bacterial aetiologies. Nevertheless, Chagas disease was the most commonly studied protozoan cause of CM. Serological tests mainly ELISA, indirect hemagglutination, and indirect immunofluorescence assays remain the test of choice for detecting protozoal infection and were the methods used in the individual studies for identifying infected (seropositive) patients and distinguishing them from health controls (seronegative individuals). Overall, the findings indicate that about a third (30%) of symptomatic individuals seropositive for protozoan cardiac infection have significant ECG abnormalities, which is 2.6 fold higher prevalence compared to healthy seronegative patients. Typical ECG abnormalities in seropositive individuals include ST-T changes, AV block, LV hypertrophy, low QRS voltage, bundle branch block, rhythm disturbances or ectopic – atrial fibrillation/flutter, bradycardia, and sinus tachycardia. However, echocardiography defined cardiac abnormalities was less common, found described only in 5.3% of seropositive patients. Male sex and older age are more likely to develop cardiac dysfunction following protozoan infection of the heart. Factors associated with poor prognosis in protozoan CM include LV systolic dysfunction, increased LV dimensions, early isovolumic relaxation, and indexed left atrial volume.
The present findings support previous review articles and expert consensus guidelines on the diagnosis and clinical management for infectious cardiomyopathies. The bulk of the studies investigated Chagasic CM consistent with reports that Chagas disease is the both the most prevalent and most studied aetiology of parasitic (protozoan) CM. The 2016 AHA scientific statement on the current diagnostic and treatment strategies for specific dilated CM recognizes Chagasic CM as an important cause of dilated CM in Central and South America, where Chagas disease is endemic but relatively uncommon in other parts of the world. [20]. Serological tests are useful in demonstrating the presence of protozoan infection as well as the causative protozoan parasite and were the choice tests for classifying patients into seropositive and seronegative arms in the included studies prior to ECG or non-invasive imaging tests. The heart is a target of protozoan infection, which affects the cardiac conduction system manifesting as ECG abnormalities: bundle branch block and AV block as well as rhythm disturbances such as atrial fibrillation or flutter, tachycardia or bradycardia, and thromboembolism over time. Death is usually a consequence of rhythm disturbances or progressive HF with complete heart block, LBBB, complex ventricular ectopy suggesting poor prognosis.
The prevalence of conduction abnormalities in protozoan CM is consistent with proposed pathophysiological mechanisms suggesting the involvement of conduction and electrical abnormalities. In addition, the findings are consistent with recommendations of both the AHA [20] and the Japanese Circulation Society Working Group for the diagnosis and treatment of myocarditis [237]. Due to a high prevalence of conduction abnormalities, the guidelines recommend initial and continuous ECG tests as a sensitive and convenient diagnostic test. ECG manifestations during the course of protozoan CM are diverse usually depending on the causative protozoan or on the natural course of the disease. Frequent ECG changes may include AV block, intraventricular conduction delay (widened QRS complex), reduced R-Wave height, abnormal Q waves, ST-T segment changes, low voltage, premature beats, tachycardia, atrial fibrillation, sinus arrest, and asystole. Continuous ECG monitoring is crucial to detect potentially fatal arrhythmias [237,238]. In a systematic review of ECG diagnosis in patients with Chagasic CM, Rojas et al. [239] finds low sensitivity, specificity, positive predictive value and negative predictive value are 40%, 76%, 48% and 69% respectively which improves significantly when more than one ECG features is tested. However, in patients with CM due to P. brucei infection (African Trypanosoma) cardiac involvement documented by ECG changes are frequently observed but of rare clinical relevance [220]. Therefore, ECG monitoring may not be required in patients with uncomplicated falciparum malaria and the lack of evidence of underlying cardiac condition [171].
In addition to ECG, tests for the levels of cardiac biomarkers are used routinely to provide complementary evidence for cardiac dysfunction to support diagnosis for coronary syndromes, myocarditis and CM. Myocardial constitutive proteins such as cardiac troponin T and creatinine kinase-MB detected in serum are often elevated suggesting myocardial injury [237]. However, the clinical value of cardiac biomarkers varies based on the primary protozoan infection. In patients with CM due to plasmodium falciparum (malaria), elevated cardiac troponin T levels is a rare event [171]. Other potential causes of elevated cardiac biomarkers such as trauma, septic shock, malignancy or cirrhosis should be evaluated and excluded to support diagnosis of CM [171]. The present findings also reveal echocardiography is a widely used test for evaluating cardiac function and morphology in patients suspected with protozoan CM. Typical echocardiographic features such as transient ventricular wall thickening, reduced wall motion and reduced cardiac chamber size in addition to pericardial effusion support the diagnosis of CM [237]. The AHA indicates cardiac infection is common in Chagas disease usually including pathological changes such as bi-ventricular hypertrophy, thinning of ventricular walls, apical aneurysm and mural thrombi. In patients with Chagasic CM, TDI septal e’ and septal E/e’ ration are useful for detecting diastolic dysfunction [232]. ECG abnormalities have also been associated with progressively higher LV dimensions (LVEDD) and depressed LV systolic function (LVEF) [229]. Finally, although cardiac magnetic resonance imaging especially T1- and T2-weighted, and late gadolinium enhanced imaging of the heart are useful to make a positive diagnosis, routine use of CMR is not widespread in clinical settings.
Clinical implications
Parasitic CM due to protozoan infection are less common relative to virus and bacteria. Protozoan CM is usually reported in areas endemic for protozoan infection such as Central and South America, and Africa. In such endemic areas, diagnosis of CM should include tests for protozoan parasites when no other causes of myocardial dysfunction have been demonstrated. Diagnosis of protozoan CM should be considered in patients presenting with cardiac symptoms of heart failure, chest pain due to pericardial irritation and symptom associated with heart block and arrhythmias. Serological testing in suspected patients is advised for causal diagnosis. Early diagnosis is crucial since cardia dysfunction in protozoan CM are transient and potentially reversible. Additional studies investigating protozoan CM is warranted to improve early detection of the causative protozoan parasite as well as the identification of at risk patients to improve diagnosis and clinical management. Additional studies are also important to understand the epidemiology of infectious CM since the predominant causative microorganism seems to change over time and across regions. The presentation of protozoan CM also varies ranging from minor symptoms of malaise due to decompensated HF, chest pain of sudden cardiac death. With increasing use of diagnostic tests such as echocardiography and CMR imaging, their value in the diagnosis and prognosis of patients with protozoan CM should be evaluated. Finally, medication for protozoan infection is advised for asymptomatic patients whereas the traditional HF medication is recommended for symptomatic patients with evidence of cardiac dysfunction, similar to medical therapies used in the management of patients diagnosed with other forms of infectious dilated CM.
Parasitic infections by protozoan can result in a wide range of clinical manifestations in humans including cardiac involvement resulting into dilated cardiomyopathy. Frequently, protozoa such as Trypanosoma cruzi, Trypanosoma brucei, toxoplasma, plasmodium and Leishmania have been implicated as a significant cause of cardiomyopathy that is more prevalent in resource-constrained countries and in immunodeficient individuals. In endemic areas, differential diagnosis for protozoan infection especially of the myocardial and pericardial diseases of unknown aetiologies should be considered in both immunocompetent and immunodeficient individuals presenting with signs and symptoms of cardiac dysfunction. Clinical presentation of protozoan cardiomyopathy varies widely due to heterogeneity of the causative parasite and thus less effective to support diagnosis. Microscopic and serological tests may identify the causative parasite and together with demonstrated evidence of conduction abnormalities, using ECG (although non-specific) and evidence of myocardial dysfunction using non-invasive imaging often echocardiography and/or CMR are useful tools to support diagnosis. Clinical management often targets treatment and elimination of the causative parasite, which often has been associated with the reversal of myocardial dysfunction and symptom resolution, which usually avoids the need for heart failure therapy in most of these patients.
- Cubillos-Garzón LA, Casas JP, Morillo CA, Bautista LE (2004) Congestive heart failure in Latin America: the next epidemic. Am Heart J 147: 412-417. [Crossref]
- Barsoum RS (2004) Parasitic infections in organ transplantation. Exp Clin Transplant 2: 258-267. [Crossref]
- Fauci AS (2001) Infectious diseases: considerations for the 21st century. Clin Infect Dis 32: 675-685. [Crossref]
- Dias JC, Silveira AC, Schofield CJ (2002) The impact of Chagas disease control in Latin America: a review. Mem Inst Oswaldo Cruz 97: 603-612. [Crossref]
- World Health Organization (2008) The global burden of disease: 2004 update: Projections of mortality and burden of disease, 2004-2030. Available at https://www.who.int/healthinfo/global_burden_disease/projections2004/en/
- Moolani Y, Bukhman G, Hotez PJ (2012) Neglected Tropical Diseases as Hidden Causes of Cardiovascular Disease. PLoS Negl Trop Dis 6: e1499. [Crossref]
- Franco‐Paredes C, Rouphael N, Méndez J, Folch E, Rodraíguez‐Morales AJ (2007) Cardiac manifestations of parasitic infections part 1: overview and immunopathogenesis. Clin Cardiol 30: 195-199. [Crossref]
- Groom ZC, Protopapas AD, Zochios V (2017) Tropical diseases of the myocardium: a review. Int J Gen Med 10: 101-111. [Crossref]
- Hidron A, Vogenthaler N, Santos-Preciado JI, Rodriguez-Morales AJ, Franco-Paredes C et al. (2010) Cardiac involvement with parasitic infections. Clin Microbiol Rev 23: 324-349. [Crossref]
- Kirchhoff LV (1993) American trypanosomiasis (Chagas' disease): a tropical disease now in the United States. N Eng J Med 329: 639-644. [Crossref]
- Rassi Jr A, Rassi A, Little WC (2000) Chagas' heart disease. Clin Cardiol 23: 883-889. [Crossref]
- Sinha A, Grace C, Alston WK, Westenfeld F, Maguire JH (1999) African trypanosomiasis in two travelers from the United States. Clin Infect Dis 29: 840-844. [Crossref]
- Leiguarda R, Roncoroni A, Taratuto AL, Jost L, Berthier M et al. (1990) Acute CNS infection by Trypanosoma cruzi (Chagas' disease) in immunosuppressed patients. Neurology 40: 850-851. [Crossref]
- Silva N, O'Bryan L, Medeiros E, Holand H, Suleiman J, De Mendonca JS, Patronas N, Reed SG, Klein HG, Masur H, Badaro R. Trypanosoma cruzi meningoencephalitis in HIV-infected patients. J Acquir Immune Defic Syndr Hum Retrovirol 20: 342-349. [Crossref]
- Rocha A, de Meneses AC, De Meneses O, da Silva AM, Ferreira MS et al. (1994) Pathology of patients with Chagas' disease and acquired immunodeficiency syndrome. Am J Trop Med Hyg 50: 261-268. [Crossref]
- Ryning FW, Mc Leod RI, Maddox JC, Hunt S, Remington JS (1979) Probable transmission of Toxoplasma gondii by organ transplantation. Ann Intern Med 90: 47-49. [Crossref]
- Gallino A, Maggiorini M, Kiowski W, Martin X, Wunderli W et al. (1996) Toxoplasmosis in heart transplant recipients. Eur J Clin Microbiol Infect Dis 15: 389-393. [Crossref]
- De Medeiros BC, De Medeiros CR, Werner B, Loddo G, Pasquini R et al. (2001) Disseminated toxoplasmosis after bone marrow transplantation: report of 9 cases. Transpl Infect Dis 3: 24-28. [Crossref]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F et al. (2008) Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29: 270-276. [Crossref]
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D et al. (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. 113: 1807-1816. [Crossref]
- Moncayo A (2003) Chagas disease: current epidemiological trends after the interruption of vectorial and transfusional transmission in the Southern Cone countries. Inst. Oswaldo Cruz 98: 577–591. [Crossref]
- Bern C (2015) Chagas’ disease. N Eng J Med 373: 456-466. [Crossref]
- Bilate A, Cunha-Neto E (2008) Chagas disease cardiomyopathy: current concepts of an old disease. Rev Inst Med Trop Sao Paulo 50: 67-74. [Crossref]
- Cunha-Neto E, Chevillard C (2014) Chagas disease cardiomyopathy: immunopathology and genetics. Mediators Inflamm 2014: 683230. [Crossref]
- Guerri-Guttenberg RA, Grana DR, Ambrosio G, Milei J (2008) Chagas cardiomyopathy: Europe is not spared! Eur Heart J 29: 2587-2591. [Crossref]
- Leiby DA, Herron RMJ, Garratty G, Herwaldt BL (2008). Trypanosoma cruzi parasitemia in US blood donors with serologic evidence of infection. J Infect Dis 198: 609-613. [Crossref]
- Laranja FS, Dias E, Nobrega G, Miranda A (1956) Chagas’ disease: a clinical, epidemiologic, and pathologic study. Circulation 14:1035-1060. [Crossref]
- Pinto DJC (1995) Natural history of Chagas disease. Arq Bras Cardiol 65:359-366. [Crossref]
- Milei J, Fernandez-Alonso G, Vanzulli S, Storino RA, Maturri L et al. (1996) Myocardial inflammatory infiltrate in human chronic Chagasic cardiomyopathy: immunohistochemical findings. Cardiovasc Pathol 5: 209-219. [Crossref]
- Nunes MC, Beaton A, Acquatella H, Bern C, Bolger AF et al. (2018) Chagas cardiomyopathy: an update of current clinical knowledge and management: a scientific statement from the American Heart Association. Circulation 138: e169-209. [Crossref]
- Rassi Jr A, Rassi A, Marin-Neto JA (2009) Chagas heart disease: pathophysiologic mechanisms, prognostic factors and risk stratification. Mem Inst Oswaldo Cruz 104: 152-158. [Crossref]
- Rassi Jr A, Rassi A, Marin-Neto JA (2010) Chagas disease. Lancet 375: 1388-1402. [Crossref]
- Factor SM, Cho S, Wittner M, Tanowitz H (1985) Abnormalities of the coronary microcirculation in acute murine Chagas’ disease. Am J Trop Med Hyg 34: 246-253. [Crossref]
- Marin-Neto JA, Cunha-Neto E, Maciel BC, Simões MV (2007) Pathogenesis of chronic Chagas heart disease. Circulation 115:1109-1123. [Crossref]
- Dutra WO, Menezes CA, Magalhaes LM, Gollob KJ (2014) Immunoregulatory networks in human Chagas disease. Parasite Immunol 36: 377-387. [Crossref]
- Bellotti G, Bocchi EA, de Moraes AV, Higuchi ML, Barbero-Marcial M et al. (1996) In vivo detection of Trypanosoma cruzi antigens in hearts of patients with chronic Chagas’ heart disease. Am Heart J 131: 301–307. [Crossref]
- Higushi ML (1993) Correlation between Trypanosoma cruzi parasitism and myocardial inflammatory infiltrate in human chronic chagasic myocarditis: light microscopy and immunohistochemical findings. Pathol 2: 101-106. [Crossref]
- Tarleton RL (2001) Parasite persistence in the aetiology of Chagas disease. Int J Parasitol 31: 550-554. [Crossref]
- Franco MF (1990) Experimental carditis induced by Trypanosoma cruzi (y strain) in guinea pigs: correlation between histopathology and the presence of cruzi antigens identified by indirect immunofluorescence. Rev Soc Bras Med Trop 23: 187-189.
- Ben YCA, Hontebeyrie-Joskowicz M, Tricottet V, Eisen H, Reynes M et al. (1988) Persistence of Trypanosoma cruzi antigens in the inflammatory lesions of chronically infected mice. Trans R Soc Trop Med Hyg 82: 77-83. [Crossref]
- Jones EM, Colley DG, Tostes S, Lopes ER, Vnencak-Jones CL et al. (1993) Amplification of a Trypanosoma cruzi DNA sequence from inflammatory lesions in human chagasic cardiomyopathy. Am J Trop Med Hyg 48: 348-357. [Crossref]
- Anez N, Carrasco H, Parada H, Crisante G, Rojas A et al. (1999) Myocardial parasite persistence in chronic chagasic patients. Am J Trop Med Hyg 60: 726-732. [Crossref]
- Zhang L, Tarleton RL (1999) Parasite persistence correlates with disease severity and localization in chronic Chagas’ disease. J Infect Dis 180: 480-486. [Crossref]
- Olivares-Villagómez D, McCurley TL, Vnencak-Jones CL, Correa-Oliveira R, Colley DG et al. (1998) Polymerase chain reaction amplification of three different Trypanosoma cruzi DNA sequences from human chagasic cardiac Am J Trop Med Hyg 59:563-570. [Crossref]
- Palomino SA, Aiello VD, Higuchi ML (2000) Systematic mapping of hearts from chronic chagasic patients: the association between the occurrence of histopathological lesions and Trypanosoma cruzi Ann Trop Med Parasitol 94: 571-579. [Crossref]
- Higuchi ML, De Morais CF, Pereira Barreto AC, Lopes EA, Stolf N et al. (1997) The role of active myocarditis in the development of heart failure in chronic Chagas’ disease: a study based on endomyocardial biopsies. Clin Cardiol 10: 665-670. [Crossref]
- Bilate AM, Salemi VM, Ramires FJ, de Brito T, Silva AM et al. (2003) The Syrian hamster as a model for the dilated cardiomyopathy of Chagas’ disease: a quantitative echocardiographical and histopathological analysis. Microbes Infect 5:1116-1124. [Crossref]
- Andrade SG, Stocker-Guerret S, Pimentel AS, Grimaud JA (1991) Reversibility of cardiac fibrosis in mice chronically infected with Trypanosoma cruzi, under specific chemotherapy. Mem Inst Oswaldo Cruz 86: 187-200. [Crossref]
- Garcia S, Ramos CO, Senra JF, Vilas-Boas F, Rodrigues MM et al. (2005) Treatment with benznidazole during the chronic phase of experimental Chagas’ disease decreases cardiac Antimicrob Agents Chemother 49: 1521-1528. [Crossref]
- Bahia MT, de Andrade IM, Martins TA, do Nascimento ÁF, Diniz Lde F et al. (2012) Fexinidazole: a potential new drug candidate for Chagas disease. PLoS Negl Trop Dis 6: e1870. [Crossref]
- Andrade ZA, Andrade SG, Sadigursky M (1987) Enhancement of chronic Tr ypanosoma cruzi myocarditis in dogs treated with low doses of cyclophosphamide. Am J Pathol 127: 467-473. [Crossref]
- Silva JS, Rossi MA (1990) Intensification of acute Trypanosoma cruzi myocarditis in BALB/c mice pretreated with low doses of cyclophosphamide or gamma J Exp Pathol 71: 33-39. [Crossref]
- Okumura M, Mester M, Iriya K, Amato Neto V, Gama-Rodrigues J (1994) Effects of immunosuppression and benzonidazole on Trypanosoma cruzi parasitism during experimental acute Chagas’ disease. Transplant Proc 26: 1587-1589. [Crossref]
- Albareda MC, Laucella SA, Alvarez MG, Armenti AH, Bertochi G et al. (2006) Trypanosoma cruzi modulates the profile of memory CD8+ T cells in chronic Chagas’ disease patients. Int Immunol 18: 465-471. [Crossref]
- Bustamante JM, Rivarola HW, Fernández AR, Enders JE, Fretes R et al.(2003) Indeterminate Chagas' disease: Trypanosoma cruzi strain and re-infection are factors involved in the progression of cardiopathy. Clin Sci (Lond) 104: 415-420. [Crossref]
- Marinho CR, Lima MR, Grisotto MG, Alvarez JM (1999) Influence of acute-phase parasite load on pathology, parasitism, and activation of the immune system at the late chronic phase of Chagas’ disease. Infect Immun 67: 308-318. [Crossref]
- Kalil J, Cunha-Neto E (1996) Autoimmunity in Chagas disease cardiomyopathy: fulfilling the criteria at last? Parasitol Today 12: 396-399. [Crossref]
- Andrade ZA (1999) Immunopathology of Chagas disease. Mem Inst Oswaldo Cruz 94: 710-80. [Crossref]
- Tarleton RL (2003) Chagas disease: a role for autoimmunity? Trends Parasitol 19: 447-451. [Crossref]
- Tarleton RL, Reithinger R, Urbina JA, Kitron U, Gürtler RE (2007) The challenges of Chagas disease: grim outlook or glimmer of hope. PLoS Med 4:e332. [Crossref]
- Kierszenbaum F (2007) Mechanisms of pathogenesis in Chagas disease. Acta Parasitol 52: 1. Doi: https://doi.org/10.2478/s11686-006-0048-y
- Bonney KM, Engman DM (2008) Chagas heart disease pathogenesis: one mechanism or many? Curr Mol Med 8: 510-518. [Crossref]
- Dutra WO, Gollob KJ. Current concepts in immunoregulation and pathology of human Chagas disease. Curr Opin Infect Dis 21: 287-292. [Crossref]
- Lopes ER, Tafuri WL (1983) Involvement of the autonomic nervous system in Chagas’ heart disease. Rev Soc Bras Med Trop 16: 206-211.
- Bohm GM (1968) Quantitative study of the intrinsic innervation of the heart in endomyocardial fibrosis and African idiopathic cardiopathies. Rev Inst Med Trop Sao Paulo 10: 84-87. [Crossref]
- Marin Neto JA, Gallo L Jr, Manco JC, Rassi A, Amorim DS (1980) Mechanisms of tachycardia on standing: studies in normal individuals and in chronic Chagas’ heart patients. Cardiovasc Res 14: 541-550. [Crossref]
- Ribeiro dos Santos R, Marquez JO, Von Gal Furtado CC, Ramos de Oliveira JC, Martins AR et al. (1979) Antibodies against neurons in chronic Chagas’ disease. Tropenmed Parasitol 30: 19-23. [Crossref]
- Ribeiro dos Santos, Hudson L (1981) Denervation and the immune response in mice infected with Trypanosoma Clin Exp Immunol 44: 349-354. [Crossref]
- Amorim DD, Marin Neto JA (1995) Functional alterations of the autonomic nervous system in Chagas’ heart Sao Paulo Med J 113: 772-784. [Crossref]
- De Alencar MC, Rocha MO, Lima MM, Costa HS, Sousa GR et al. (2014) Heart rate recovery in asymptomatic patients with Chagas disease. PLoS One 9: e100753. [Crossref]
- Marin-Neto JA, Bromberg-Marin G, Pazin-Filho A, Simões MV, Maciel BC (1998) Cardiac autonomic impairment and early myocardial damage involving the right ventricle are independent phenomena in Chagas’ disease. Int J Cardiol 65: 261-269. [Crossref]
- Ribeiro AL, Moraes RS, Ribeiro JP, Ferlin EL, Torres RM et al. (2001) Parasympathetic dysautonomia precedes left ventricular systolic dysfunction in Chagas disease. Am Heart J 2001;141: 260-265. [Crossref]
- Sousa AC, Marin-Neto JA, Maciel BC, Gallo L Jr, Amorim DS (1987) Cardiac parasympathetic impairment in gastrointestinal Chagas’ disease. Lancet 1: 985. [Crossref]
- Koberle F (1968) Chagas’ disease and Chagas’ syndromes: the pathology of American trypanosomiasis. Adv Parasitol 6: 63-116. [Crossref]
- Cuba MB, Machado MP, Farnesi TS, Alves AC, Martins LA et al. (2014) Effects of cholinergic stimulation with pyridostigmine bromide on chronic chagasic cardiomyopathic Mediators Inflamm 2014: 475946. [Crossref]
- Andrade ZA, Andrade SG, Sadigursky M, Wenthold RJ Jr, Hilbert SL et al. (1997) The indeterminate phase of Chagas’ disease: ultrastructural characterization of cardiac changes in the canine model. Am J Trop Med Hyg 57: 328-336. [Crossref]
- Rabelo DR, Rocha MO, de Barros MV, Silva JL, Tan TC, Nunes MC (2014) Impaired coronary flow reserve in patients with indeterminate form of Chagas’ Echocardiography 31: 67-73. [Crossref]
- Oliveira JS (1985) A natural human model of intrinsic heart nervous system denervation: Chagas’ cardiopathy. Am Heart J 110: 1092-1098. [Crossref]
- Miranda CH, Figueiredo AB, Maciel BC, Marin-Neto JA, Simões MV (2011) Sustained ventricular tachycardia is associated with regional myocardial sympathetic denervation assessed with 123I-metaiodobenzylguanidine in chronic Chagas cardiomyopathy. J Nucl Med 52: 504-510. [Crossref]
- Miranda CH, Gadioli LP, Pintya AO, Figueiredo AB, Maciel BC et al. (2018) The severity of ventricular arrhythmia correlates with the extent of myocardial sympathetic denervation, but not with myocardial fibrosis extent in chronic Chagas cardiomyopathy. J Nucl Cardiol 25: 75-83. [Crossref]
- Hagar JM, Rahimtoola SH (1991) Chagas’ heart disease in the United States. N Engl J Med 325: 763-768. [Crossref]
- Marin-Neto JA, Marzullo P, Marcassa C, Gallo L Jr, Maciel BC et al. (1992) Myocardial perfusion abnormalities in chronic Chagas’ disease as detected by thallium-201 scintigraphy. Am J Cardiol 69: 780-784. [Crossref]
- Rossi MA, Tanowitz HB, Malvestio LM, Celes MR, Campos EC et al. (2010) Coronary microvascular disease in chronic Chagas cardiomyopathy including an overview on history, pathology, and other proposed pathogenic PLoS Negl Trop Dis 4: e674. [Crossref]
- Andrade ZA, Andrade SG, Correa R, Sadigursky M, Ferrans VJ (1994) Myocardial changes in acute Trypanosoma cruzi infection: ultrastructural evidence of immune damage and the role of microangiopathy. Am J Pathol 144: 1403-1411. [Crossref]
- Ferrans VJ, Milei J, Tomita Y, Storino RA (1988) Basement membrane thickening in cardiac myocytes and capillaries in chronic Chagas’ disease. Am J Cardiol 61: 1137-1140. [Crossref]
- Hiss FC, Lascala TF, Maciel BC, Marin-Neto JA, Simões MV (2009) Changes in myocardial perfusion correlate with deterioration of left ventricular systolic function in chronic Chagas’ cardiomyopathy. JACC Cardiovasc Imaging 2: 164-172. [Crossref]
- Andrade JP, Marin Neto JA, Paola AA, Vilas-Boas F, Oliveira GM et al. (2011) Latin American guidelines for the diagnosis and treatment of Chagas’ heart disease: executive summary. Arq Bras Cardiol 96: 434-444. [Crossref]
- Maguire JH, Hoff R, Sherlock IT, Guimarães AC, Sleigh AC et al. (1987) Cardiac morbidity and mortality due to Chagas' disease: prospective electrocardiographic study of a Brazilian community. Circulation 75: 1140-1145. [Crossref]
- Migliore RA, Guerrero FT, Armenti A, Fernández C, Adaniya ME et al. (1990) Diastolic function in Chagas disease. Medicina 50: 537-542. [Crossref]
- Acquatella H (2007) Echocardiography in Chagas heart disease. Circulation 115: 1124-1131. [Crossref]
- Rassi Jr A, Rassi A, Rassi SG (2007) Predictors of mortality in chronic Chagas disease: a systematic review of observational studies. Circulation 115: 1101-1108. [Crossref]
- Carrasco HA, Parada H, Guerrero L, Duque M, Durán D et al. (1994) Prognostic implications of clinical, electrocardiographic and hemodynamic findings in chronic Chagas' disease. Int J Cardiol 43: 27-38. [Crossref]
- Espinosa R, Carrasco HA, Belandria F, Fuenmayor AM, Molina C et al. (1985) Life expectancy analysis in patients with Chagas' disease: prognosis after one decade (1973–1983). Int J Cardiol 8: 45-56. [Crossref]
- Freitas HF, Chizzola PR, Paes ÂT, Lima AC, Mansur AJ (2005) Risk stratification in a Brazilian hospital-based cohort of 1220 outpatients with heart failure: role of Chagas' heart disease. Int J Cardiol 102: 239-247. [Crossref]
- Li E, Stanley SL (1996). Protozoa. Amebiasis. Gastroenterol. Clin North Am 25: 471-492. [Crossref]
- Clumeck N (1991) Some aspects of the epidemiology of toxoplasmosis and pneumocystosis in AIDS in Europe. Eur J Clin Microbiol Infect Dis 10: 177-178. [Crossref]
- Andy JJ, O'Connell JP, Daddario RC, Roberts WC (1977) Trichinosis causing extensive ventricular mural endocarditis with superimposed thrombosis: Evidence that severe eosinophilia damages endocardium. Am J Med 63: 824-829. [Crossref]
- Jones TC, Yeh S, Hirsch JG (1972) The interaction between Toxoplasma gondii and mammalian cells: I. Mechanism of entry and intracellular fate of the parasite. J Exp 136: 1157-1172. [Crossref]
- Adair OV, Randive N, Krasnow N (1989) Isolated toxoplasma myocarditis in acquired immune deficiency syndrome. Am Heart J 118: 856-857. [Crossref]
- Samuel J, Oliveira M, Correa DA, Navarro MA, Muccillo G (1983) Cardiac thrombosis and thromboembolism in chronic Chagas' heart disease. Am J Cardiol 52: 147-151. [Crossref]
- Rassi Jr A, Rassi SG, Rassi A (2001) Sudden death in Chagas' disease. Arq Bras Cardiol 76: 86-96. [Crossref]
- Carod-Artal FJ, Vargas AP, Horan TA, Nunes LG (2005) Chagasic cardiomyopathy is independently associated with ischemic stroke in Chagas disease. Stroke 36: 965-970. [Crossref]
- World Health Organization (2002) Control of Chagas disease. World Health Organ Tech Rep Ser 905: 1-10. [Crossref]
- Tanowitz HB, Machado FS, Jelicks LA, Shirani J, de Carvalho AC et al. (2009) Perspectives on Trypanosoma cruzi–induced heart disease (Chagas disease). Prog Cardiovasc Dis 51: 524-539. [Crossref]
- Viotti RJ, Vigliano C, Laucella S, Lococo B, Petti M et al. (2004) Value of echocardiography for diagnosis and prognosis of chronic Chagas cardiomyopathy without heart failure. Heart 90: 655-660. [Crossref]
- Gourbière S, Dumontiel E, Rabinovich JE, Minkoue R, Menu F (2008) Demographic and dispersal constraints for domestic infestation by nondomicilated Chagas disease vectors in the Yucatan Peninsula, Am J Trop Med Hyg 78: 133-139. [Crossref]
- Ceaser M (2005) Battling with Chagas disease in South America. Lancet Infect Dis 5: 470-471.
- Simarro PP, Jannin J, Cattand P (2008) Eliminating human African trypanosomiasis: where do we stand and what comes next? PLoS Med 5: e55. [Crossref]
- Blum J, Schmid C, Burri C. (2006) Clinical aspects of 2541 patients with second stage human African trypanosomiasis. Acta Trop 97: 55-64. [Crossref]
- Morrison WI, Murray M, Sayer PD, Preston JM (1981) The pathogenesis of experimentally induced Trypanosoma brucei infection in the dog: II. Changes in the lymphoid organs. Am J Pathol 102: 168-181. [Crossref]
- Poltera AA, Hochmann A, Lambert PH (1980) A model for cardiopathy induced by Trypanosoma brucei brucei in mice. A histologic and immunopathologic study. Am J Pathol 99: 325-352. [Crossref]
- Poltera AA, Sayer PD, Rudin W, Bovell D (1985) Trypanosomal cardiac valvulitis in vervet monkeys. Trop Med Parasitol 36: 77-80. [Crossref]
- Adams JH, Haller L, Boa FY, Doua F, Dago A et al. (1986) Human African trypanosomiasis (Tb gambiense): a study of 16 fatal cases of sleeping sickness with some observations on acute reactive arsenical encephalopathy. Neuropathol Appl Neurobiol 12: 81-94. [Crossref]
- Poltera AA, Owor R, Cox JN (1977) Pathological aspects of human African trypanosomiasis (HAT) in Uganda. Virchows Arch A Pathol Anat Histol 373: 249-265. [Crossref]
- Koten JW, De Raadt P (1969) Myocarditis in Trypanosoma rhodesiense infections. Trans R Soc Trop Med Hyg 63: 485-489. [Crossref]
- De Raadt P, Koten JW (1968) Myocarditis in Rhodesiense trypanosomiasis. East Afr Med J 45: 128-132. [Crossref]
- Blum JA, Burri C, Hatz C, Kazumba L, Mangoni P et al. (2007) Sleeping hearts: the role of the heart in sleeping sickness (human African trypanosomiasis). Trop Med Int Health 12: 1422-1432. [Crossref]
- Bertrand E, Serie F, Kone I, Rive J, Campaore P et al. (1973) Symptomatologie générale de la trypanosomiase humaine africaine au moment du dépistage. Med Afr Noire 20: 303-314.
- Brun R, Blum J, Chappuis F, Burri C (2010) Human African trypanosomiasis. Lancet 375: 148-159. [Crossref]
- Bailey JW, Smith DH (1994) The quantitative buffy coat for the diagnosis of trypanosomes. Trop Doct 24: 54-56. [Crossref]
- Miezan TW, Meda AH, Doua F, Cattand P (1990) Evaluation of the parasitologic technics used in the diagnosis of human Trypanosoma gambiense trypanosomiasis in the Ivory Coast. Bull Soc Pathol Exot 87: 101-104. [Crossref]
- WHO Expert Committee on the Control, Surveillance of African Trypanosomiasis, World Health Organization (1998) Control and Surveillance of African Trypanosomiasis: Report of a WHO Expert Committee. World Health Organ Tech Rep Ser 1-114. [Crossref]
- Collomb H, Bartoli D. The heart in human African trypanosomiasis caused by Trypanosoma gambiense. Bull Soc Pathol Exot Filiales 60: 142-146. [Crossref]
- Tsala PM, Blackett K, Mbonifor CL, Leke R, Etoundi J (1998) Functional and immunologic involvement in human African trypanosomiasis caused by Trypanosoma gambiense. Bull Soc Pathol Exot Filiales 81: 490-501. [Crossref]
- Checchi F, Filipe JA, Barrett MP, Chandramohan D (2008) The natural progression of Gambiense sleeping sickness: what is the evidence? PLoS Negl Trop Dis 2: e303. [Crossref]
- WHO (2003) WHO report on African trypanosomiasis (sleeping sickness). Report of the Scientific Working Group meeting on African trypanosomiasis Geneva, 4-8 June, 2001. World Health Organization, Geneva, Switzerland
- Krick JA, Remington JS (1978) Toxoplasmosis in the adult: an overview. N Eng J Med 298: 550-553. [Crossref]
- Frenkel JK 1973. Toxoplasma in and around us. Bio Sci 23: 343-352.
- Herwaldt BL, Juranok DD (1993) Laboratory acquired malaria, leishmaniasis, trypanosomiasis and toxoplasmosis. Am J Trop Med Hyg 48: 313-323. [Crossref]
- Feldman HA (1974) Toxoplasmosis: an overview. Bull N Y Acad Med 50: 110-127. [Crossref]
- Ferreira MS, Borges AS (2002) Some aspects of protozoan infections in immunocompromised patients: a review. Mem Inst Oswaldo Cruz 97: 443-457. [Crossref]
- Frenkel JK (1957) Effects of cortisone, total body irradiation and nitrogen mustard on chronic latent toxoplasmosis. Am J Pathol 33: 618.
- Tschirhart D, Klatt EC (1988) Disseminated toxoplasmosis in the acquired immnunodeficiency syndrome. Arc Path Labor Med 112: 1237-1241. [Crossref]
- Jautzke G, Sell M, Thal Mann V, Janitschke K, Gottscha LK et al. (1993) Extracerebral toxoplasmosis in AIDS: histological and immunohistological findings basedn on 80 autopsy cases. Pathol Res Pract 189: 428-436. [Crossref]
- Levy J, Espanol-Boren T, Thomas C, Fischer A, Tovo P et al. (1997) Clinical spectrum of X-linked hyper-IgM syndrome. J Paediatr 131: 47-54. [Crossref]
- Subauste CS, Wessendarp M, Smulian AG, Frame PT (2001) Role of CD40 ligand signaling in defective type 1 cytokine response in human immunodeficiency virus infection. J Infect Dis 183: 1722-1731. [Crossref]
- Brown CR, McLeod R (1990) Class I MHC genes and CD8+ T cells determine cyst number in Toxoplasma gondii infection. J Immunol 145: 3438-3441. [Crossref]
- Suzuki Y, Wong SY, Grumet FC, Fessel J, Montoya JG et al. (1996) Evidence for genetic regulation of susceptibility to toxoplasmic encephalitis in AIDS patients. J Infect Dis 173: 265-268. [Crossref]
- Mack DG, Johnson JJ, Roberts F, Roberts CW, Estes RG et al. (1999) HLA-class II genes modify outcome of Toxoplasma gondii infection. Int J Parasitol 29: 13511-1358. [Crossref]
- Hofman P, Bernard E, Michiels JF, Thyss A, Le Fichoux Y et al. (1993) Extracerebral toxoplasmosis in the acquired immunodeficiency syndrome (AIDS). Pathol Res Pract 189: 894-901. [Crossref]
- Politi MO, Rubens MM (1996) Pathology of the heart in AIDS. Study of 73 consecutive necropsies. Arq Bras Cardiol66: 129-133. [Crossref]
- Sahasrabudhe NS, Jadhav MV, Deshmukh SD, Holla VV (2003) Pathology of Toxoplasma myocarditis in acquired immunodeficiency syndrome. Indian J Pathol Microbiol 46: 649-651. [Crossref]
- Wagner FM, Reichenspurner H, Uberfuhr P, Weiss M, Fingerle V et al. (1994) Toxoplasmosis after heart transplantation: diagnosis by endomyocardial biopsy. J Heart Lung Transplant 13: 916-918. [Crossref]
- Mayes JT, O'Connor BJ, Avery R, Castellani W, Carey W (1995) Transmission of Toxoplasma gondii infection by liver transplantation. Clin Infect Dis 21: 511-515. [Crossref]
- Reynolds ES, Walls KW, Pfeiffer RI (1966) Generalized toxoplasmosis following renal transplantation: report of a case. Arch Intern Med 118: 401-405. [Crossref]
- Slavin MA, Meyers JD, Remington JS, Hackman RC (1994) Toxoplasma gondii infection in marrow transplant recipients: a 20 year experience. Bone Marrow Transplant 13: 549-557. [Crossref]
- Wreghitt TG, Gray JJ, Pavel P, Balfour A, Fabbri A et al. (1992) Efficacy of pyrimethamine for the prevention of donor-acquired Toxoplasma gondii infection in heart and heart-lung transplant patients. Transpl Int. 1992 5: 197-200. [Crossref]
- Anderson SE, Remington JS (1975) The diagnosis of toxoplasmosis. South Med J 68: 1433-1443. [Crossref]
- Adams JL (1962) Acute Toxoplasmosis with involment of the Heart. N Z Med J 61: 20-24. [Crossref]
- Kean BH, Grocott RG (1945) Sarcosporidiosis or toxoplasmosis in man and guinea-pig. Am J Pathol 21: 467-483. [Crossref]
- Hermanns B, Brunn A, Schwarz ER, Sachweh JS, Seipelt I et al. (2001) Fulminant toxoplasmosis in a heart transplant recipient. Pathol Res Pract 197: 211-215. [Crossref]
- Luft BJ, Billingham M, Remington JS (1986) Endomyocardial biopsy in the diagnosis of toxoplasmic myocarditis. Transplant Proc 18: 1871-1873. [Crossref]
- Roldan EO, Moskowitz L, Hensley GT (1987) Pathology of the heart in acquired immunodeficiency syndrome. Arch Pathol Lab Med 111: 943-946. [Crossref]
- Holliman R, Johnson J, Savva D, Cary N, Wreghitt T (1992) Diagnosis of toxoplasma infection in cardiac transplant recipients using the polymerase chain reaction. J Clin Pathol 45: 931-932. [Crossref]
- Mofenson LM, Oleske J, Serchuck L, Van RD, Wilfert C (2004) Treating opportunistic infections among HIV-exposed and infected children: recommendations from CDC, the National Institutes of Health, and the Infectious Diseases Society of America. MMWR Recomm Rep 53: 1-92. [Crossref]
- Derouin F, Pelloux H, ESCMID Study Group on Clinical Parasitology (2008) Prevention of toxoplasmosis in transplant patients. Clin Microbiol Infect 14: 1089-1101. [Crossref]
- Mishra SK, Behera PK, Satpathi S (2013) Cardiac involvement in malaria: an overlooked important complication. J Vector Borne Dis 50: 232. [Crossref]
- Marrelli MT, Brotto M (2016) The effect of malaria and anti-malarial drugs on skeletal and cardiac muscles. Malar J 15: 524. [Crossref]
- Ray HN, Doshi D, Rajan A, Singh AK, Singh SB et al. (2017) Cardiovascular involvement in severe malaria: A prospective study in Ranchi, Jharkhand. J Vector Borne Dis 54: 177-182. [Crossref]
- Nayak KC, Meena SL, Gupta BK, Kumar S, Pareek V (2013) Cardiovascular involvement in severe vivax and falciparum malaria. J Vector Borne Dis 50: 285. [Crossref]
- Costenaro P, Benedetti P, Facchin C, Mengoli C, Pellizzer G (2011) Fatal myocarditis in course of Plasmodium falciparum infection: case report and review of cardiac complications in malaria. Case Rep Med 2011: 202083. [Crossref]
- World Health Organization (2000) Communicable Diseases Cluster. Severe falciparum malaria. Trans R Soc Trop Med Hyg 94: S1–90. [Crossref]
- Ehrhardt S, Mockenhaupt FP, Anemana SD, Otchwemah RN, Wichmann D et al. (2005) High levels of circulating cardiac proteins indicate cardiac impairment in African children with severe Plasmodium falciparum malaria. Microbe Infect 7: 1204–1210. [Crossref]
- Herr J, Mehrfar P, Schmiedel S, Wichmann D, Brattig NW et al. (2011) Reduced cardiac output in imported Plasmodium falciparum malaria. Malar J 10: 160. [Crossref]
- Janka JJ, Koita OA, Traoré B, Traoré JM, Mzayek F et al. (2010) Increased pulmonary pressures and myocardial wall stress in children with severe malaria. J Infect Dis 202: 791-800. [Crossref]
- Costenaro P, Benedetti P, Facchin C, Mengoli C, Pellizzer G (2011) Fatal myocarditis in course of Plasmodium falciparum infection: case report and review of cardiac complications in malaria. Case Rep Med. [Crossref]
- Stout TT, van Bergeijk L, Bouma HG, Woittiez AJ (1990) Electrocardiographic changes in acute malaria. Neth J Med 37: 124-128. [Crossref]
- Franzen D, Curtius JM, Heitz W, Höpp HW, Diehl V et al. (1992) Cardiac involvement during and after malaria. Clin Investig 70: 670-673. [Crossref]
- Bethell DB, Phuong PT, Phuong CX, Nosten F, Waller D et al. (1996) Electrocardiographic monitoring in severe falciparum malaria. Trans R Soc Trop Med Hyg 90: 266-269. [Crossref]
- Gunther A, Grobusch MP, Slevogt H, Abel W, Burchard GD (2003) Myocardial damage in falciparum malaria detectable by cardiac troponin T is rare. Trop Med Int Health 8: 30-32. [Crossref]
- Ehrhardt S, Wichmann D, Hemmer CJ, Burchard GD, Brattig NW (2004) Circulating concentrations of cardiac proteins in complicated and uncomplicated Plasmodium falciparum malaria. Trop Med Int Health 9: 1099-1103. [Crossref]
- Ehrhardt S, Mockenhaupt FP, Anemana SD, Otchwemah RN, Wichmann D et al. (2005) High levels of circulating cardiac proteins indicate cardiac impairment in African children with severe Plasmodium falciparum malaria. Microbe Infect 7: 1204-1210. [Crossref]
- Lang K, Borner A, Figulla H (2000) Comparision of biochemical markers for the detection of minimal myocardial injury: superior sensitivity of cardiac troponin-T ELISA. J Int Med 247: 119-123. [Crossref]
- Aviles RJ, Askari AT, Lindahl B, Wallentin L, Jia G et al. (2002) Troponin T levels in patients with acute coronary syndromes, with or without renal dysfunction. N Engl J Med 27: 2047-2052. [Crossref]
- Wennicke K, Debierre-Grockiego F, Wichmann D, Brattig NW, Pankuweit S et al. (2008) Glycosylphosphatidylinositol-induced cardiac myocyte death might contribute to the fatal outcome of Plasmodium falciparum malaria. Apoptosis 13: 857-866. [Crossref]
- Bhat S, Alva J, Muralidhara K, Fahad S (2012) Malaria and the heart. BMJ Case Rep 2012: bcr2012007275. [Crossref]
- Bhat S, Kumar M, Alva J (2013) Malaria and the conducting system of the heart. BMJ Case Rep 2013: bcr2012007462. [Crossref]
- Dev N, Gadpayle AK, Sankar J, Choudhary M (2014) An unusual case of heart failure due to Plasmodium vivax infection with a favorable outcome. Rev Soc Bras Med Trop 47: 663-665. [Crossref]
- Vuong PN, Richard F, Snounou G, Coquelin F, Rénia L et al. (1999) Development of irreversible lesions in the brain, heart and kidney following acute and chronic murine malaria infection. Parasitol 119: 543-553. [Crossref]
- McGwire BS, Satoskar AR (2013) Leishmaniasis: clinical syndromes and treatment. QJM: Int J Med 107: 7-14. [Crossref]
- Armin S, Gharib A, Khalaj E (2008) Heart involvement in kala azar. Pak J Med Sci 24: 879-882.
- Expert Committee (2010) Control of the Leishmaniases. WHO Technical Report Series 949. Geneva, Switzerland: World Health Organization, 2010: 1–186
- Chang KP, Dwyer DM (1976) Multiplication of a human parasite (Leishmania donovani) in phagolysosomes of hamster macrophages in vitro. Science 193: 678-680. [Crossref]
- Liese J, Schleicher U, Bogdan C (2008) The innate immune response against Leishmania parasites. Immunobiology 213: 377-387. [Crossref]
- Chang KP, Reed SG, McGwire BS, Soong L (2003) Leishmania model for microbial virulence: the relevance of parasite multiplication and pathoantigenicity. Acta Trop 85: 375-390. [Crossref]
- Lopez-Pena,M, Aleman N, Munoz F, Fondevila D, Suárez ML et al. (2009). Visceral leishmaniasis with cardiac involvement in a dog: a case report. Acta Vet Scand 51: 20 [Crossref]
- Shrivastava R, Sinha PR, Singh VP, Sundar S (2007) Echocardiographic evaluation of cardiac status in Indian visceral leishmaniasis patients. Trans R Soc Trop Med Hyg 101: 429-432. [Crossref]
- Kirchhoff LV, Weiss LM, Wittner M, Tanowitz HB (2004) Parasitic diseases of the heart. Front Biosci 9: 706-723. [Crossref]
- Gilles H, Galea W (2000) Parasitic disease affecting the heart in childhood. Images Paediatr Cardiol 2: 28-39. [Crossref]
- Ravdin JI (1995) Amebiasis. Infect. Dis. 20: 1453-1464. [Crossref]
- Supe AN, Sathe SS, Redkar RG, Dalvi AN, Kulkarni BA et al. (1991) Amebic pericarditis following ruptured right liver lobe abscess. Indian J Gastroenterol 10: 111. [Crossref]
- Campbell D, Chadee K (1997) Survival strategies of Entamoeba histolytica: modulation of cell-mediated immune responses. Parasitology Today 13: 184-189. [Crossref]
- Haque R, Duggal P, Ali IM, Hossain MB, Mondal D et al. (2002) Innate and acquired resistance to amebiasis in Bangladeshi children. Infect Dis 186: 547-552. [Crossref]
- Haque R, Mondal D, Duggal P, Kabir M, Roy S et al. (2006) Entamoeba histolytica infection in children and protection from subsequent amebiasis. Infect Immun 74: 904-909. [Crossref]
- Lidell ME, Moncada DM, Chadee K, Hansson GC (2006) Entamoeba histolytica cysteine proteases cleave the MUC2 mucin in its C-terminal domain and dissolve the protective colonic mucus gel. Proc Natl Acad Sci 103: 9298-9303. [Crossref]
- Mirelman D, Anbar M, Bracha R (2008) Trophozoites of Entamoeba histolytica epigenetically silenced in several genes are virulence-attenuated. Parasite (Paris, France). 15: 266-274. [Crossref]
- Winkelmann J, Leippe M, Bruhn H (2006) A novel saposin-like protein of Entamoeba histolytica with membrane-fusogenic activity. Mol Biochem Parasitol 147: 85-94. [Crossref]
- Kulpati DD, Gupta GD, Chatrath UC, Bhargava SP, Sharma ML (1966) Amoebic pericarditis. J Assoc Physicians India 14: 679. [Crossref]
- Kulpati DD, Venkatachulam CG, Saha NC (1976) Amoebic pericarditis-a case report. J Assoc Physicians India 24: 119-122. [Crossref]
- Heller RF, Gorbach SL, Tatooles CJ, Loeb HS, Rahimtoola SH (1972) Amebic pericarditis. JAMA 220: 988-990. [Crossref]
- Mondragon-Sanchez R, Cortes-Espinoza T, Sanchez-Cisneros R, Parra-Silva H, Hurtado-Andrade H (1994) Rupture of an amebic liver abscess into the pericardium. Presentation of a case and review of current management. Hepato-gastroenterology 41: 585-588. [Crossref]
- Aikat BK, Bhusnurmath SR, Pal AK, Chhuttani PN, Datta DV (1978) Amoebic liver abscess-a clinicopathological study. Indian J Med Res 67: 381-391. [Crossref]
- Singh A, Houpt E, Petri WA (2009) Rapid diagnosis of intestinal parasitic protozoa, with a focus on Entamoeba histolytica. Interdiscip Perspect Infect Dis 547090. [Crossref]
- Zengzhu G, Bracha R, Nuchamowitz Y, Cheng W, Mirelman D (1999) Analysis by Enzyme-Linked Immunosorbent Assay and PCR of Human Liver Abscess Aspirates from Patients in China forEntamoeba histolytica. J Clin Microbiol 37: 3034-3036. [Crossref]
- Shamsuzzaman SM, Hashiguchi Y (2002) Thoracic amebiasis. Clin Chest Med 23: 479-492. [Crossref]
- Cameron EW (1978) The treatment of pleuropulmonary amebiasis with metronidazole. Chest 73: 647-650. [Crossref]
- Schuster FL, Ramirez-Avila L (2008) Current world status of Balantidium coli. Clin Microbiol Rev 21: 626-638. [Crossref]
- Areán VM, Koppisch E (1956) Balantidiasis: a review and report of cases. Am J Pathol 32: 1089. [Crossref]
- Gupta S, Bharati P, Sinha KP (2017) Balantidium Coli: Rare Urinary Pathogen or Faecal Contaminant in Urine? Case Study and Review. Journal of Dental and Medical Sciences 16: 88-90.
- Bel GS, Couret M (1910) Balantidium coli infection in man. J Infect Dis 25 :609-624.
- Fayer R (2004) Sarcocystis spp. in human infections. Clin Microbiol Rev 17: 894-902. [Crossref]
- Levine ND (1986) The taxonomy of Sarcocystis (protozoa, apicomplexa) species. J Parasitol 1: 372-382. [Crossref]
- Sosa-Jurado F, Mazariego-Aranda M, Hernández-Becerril N, Garza-Murillo V, Cárdenas M et al. (2003) Electrocardiographic findings in Mexican chagasic subjects living in high and low endemic regions of Trypanosoma cruzi infection. Mem Inst Oswaldo Cruz 98: 605-610. [Crossref]
- Angel M, Villar JC, Herrera M (2004) Electrocardiographic study in subjects with positive and negative serology for Trypanosoma Cruzi-asymptomatic patients coming from the Chicamocha study (cardiovascular health investigation from Colombia to assess the markers and outcomes of Chagas Disease). Revista Colombiana de Cardiologia 11: 246-250.
- Goldbaum M, Ajimura FY, Litvoc J, Carvalho SA, Eluf-Neto J (2004). American trypanosomiasis and electrocardiographic alterations among industrial workers in Sao Paulo, Brazil. Revista do Instituto de Medicina Tropical de São Paulo 46: 299-302. [Crossref]
- Becerril-Flores MA, Rangel-Flores E, Imbert-Palafox JL, Gomez-Gomez JV, Figueroa-Gutierrez AH (2007) Human infection and risk of transmission of Chagas disease in Hidalgo State, Mexico. Am J Trop Med Hyg 76: 318-323. [Crossref]
- Williams-Blangero S, Magalhaes T, Rainwater E, Blangero J, Correa-Oliveira R et al. (2007) Electrocardiographic characteristics in a population with high rates of seropositivity for Trypanosoma cruzi infection. Am J Trop Med Hyg 77: 495-499. [Crossref]
- Borges-Pereira J, Sarquis O, Zauza PL, Britto C, Lima MM (2008) Epidemiology of Chagas disease in four rural localities in Jaguaruana, State of Ceará: seroprevalence of infection, parasitemia and clinical characteristics. Rev Soc Bras Med Trop 41: 345-351. [Crossref]
- Brum-Soares LM, Xavier SS, Sousa AS, Borges-Pereira J, Ferreira JM et al. (2010) Morbidity of Chagas disease among autochthonous patients from the Rio Negro microregion, State of Amazonas. Rev Soc Bras Med Trop 43: 170-177. [Crossref]
- Moretti E, Castro I, Franceschi C, Basso B (2010) Chagas disease: serological and electrocardiographic studies in Wichi and Creole communities of Misión Nueva Pompeya, Chaco, Argentina. Mem Inst Oswaldo Cruz 105: 621-627. [Crossref]
- Silva ÉM, Rocha MO, Silva RC, Paixão GD, Buzzati H et al. (2010) Clinic and epidemiological study on Chagas disease in the Serra Azul district of Mateus Leme, central-western region of the State of Minas Gerais, Brazil. Rev Soc Bras Med Trop 43: 178-181. [Crossref]
- Tobar IB, Parra F, Pérez CN, Rodríguez-Bonfante C, Useche F et al. (2011) Prevalence of Trypanosoma cruzi antibodies and inflammatory markers in uncompensated heart failure. Rev Soc Bras Med Trop 44: 691-696. [Crossref]
- Monteon V, Alducin C, Hernández J, Ramos-Ligonio A, Lopez R (2013) High frequency of human blood in Triatoma dimidiata captured inside dwellings in a rural community in the Yucatan Peninsula, Mexico, but low antibody seroprevalence and electrocardiographic findings compatible with Chagas disease in humans. Am J Trop Med Hyg 88: 566-571. [Crossref]
- Ribeiro AL, Sabino EC, Marcolino MS, Salemi VM, Ianni BM et al. (2013) Electrocardiographic abnormalities in Trypanosoma cruzi seropositive and seronegative former blood donors. PLoS Negl Trop Dis 7: e2078. [Crossref]
- Molina-Garza ZJ, Rosales-Encina JL, Mercado-Hernández R, Molina-Garza DP, Gomez-Flores R et al. (2014) Association of Trypanosoma cruzi infection with risk factors and electrocardiographic abnormalities in northeast Mexico. BMC Infect Dis 14: 117. [Crossref]
- Ribeiro AL, Marcolino MS, Prineas RJ, Lima‐Costa MF (2014) Electrocardiographic Abnormalities in Elderly Chagas Disease Patients: 10‐Year Follow‐Up of the Bambuí Cohort Study of Aging. J Am Heart Assoc 3: e000632. [Crossref]
- Alroy KA, Huang C, Gilman RH, Quispe-Machaca VR, Marks MA et al. (2015) Prevalence and transmission of Trypanosoma cruzi in people of rural communities of the high jungle of Northern Peru. PLoS Negl Trop Dis 9: e0003779. [Crossref]
- Fernandez AB, Nunes MC, Clark EH, Samuels A, Menacho S et al. (2015) Electrocardiographic and echocardiographic abnormalities in Chagas disease: findings in residents of rural Bolivian communities hyperendemic for Chagas disease. Glob Heart 10: 159-166. [Crossref]
- Yager JE, Beltran DF, Torrico F, Gilman RH, Bern C (2015) Prevalence of Chagas heart disease in a region endemic for Trypanosoma cruzi: evidence from a central Bolivian community. Glob Heart 10: 145-150. [Crossref]
- Combellas I, Puigbo JJ, Acquatella H, Tortoledo F, Gomez JR (1985) Echocardiographic features of impaired left ventricular diastolic function in Chagas's heart disease. Br Heart J 53: 298-309. [Crossref]
- Barros MV, Machado FS, Ribeiro AL, da Costa Rocha MO (2004) Diastolic function in Chagas' disease: an echo and tissue Doppler imaging study. Eur J Echocardiogr 5: 182-188. [Crossref]
- Viotti RJ, Vigliano C, Laucella S, Lococo B, Petti M et al. (2004) Value of echocardiography for diagnosis and prognosis of chronic Chagas disease cardiomyopathy without heart failure. Heart 90: 655-660. [Crossref]
- Rassi DD, Vieira ML, Arruda AL, Hotta VT, Furtado RG et al. (2014) Echocardiographic parameters and survival in Chagas heart disease with severe systolic dysfunction. Arq Bras Cardiol 102: 245-252. [Crossref]
- Sánchez-Montalvá A, Salvador F, Rodríguez-Palomares J, Sulleiro E, Sao-Avilés A et al. (2016) Chagas cardiomyopathy: usefulness of EKG and echocardiogram in a non-endemic country. PloS one 11: e0157597. [Crossref]
- Franzen D, Curtius JM, Heitz W, Höpp HW, Diehl V et al. (1992) Cardiac involvement during and after malaria. Clin Investig 70: 670-673. [Crossref]
- JCS Joint Working Group (2011) Guidelines for diagnosis and treatment of myocarditis (JCS 2009): digest version. Circ 75: 734-743. [Crossref]
- Jefferies JL, Towbin JA (2010). Dilated cardiomyopathy. Lancet 375(9716): 752-762. [Crossref]
- Rojas LZ, Glisic M, Pletsch-Borba L, Echeverría LE, Bramer WM et al. (2018) Electrocardiographic abnormalities in Chagas disease in the general population: A systematic review and meta-analysis. PLoS Negl Trop Dis 12: e0006567. [Crossref]