Traditionally, the secondary complications of type 2 diabetes are considered to be caused by chronic hyperglycemia existing over a number of years and involve primarily microvascular disease of eyes, kidneys, and nerves as well as macrovascular disease of large arteries. The cause of progressive hyperglycemia is attributed to the continuing deterioration of pancreatic islet beta cell function. However, this formulation does not directly address why the beta cell continues to deteriorate over the course of the disease.
Oxidative stress consequent to chronic hyperglycemia accompanies the development of microvascular and macrovascular disease and is typically cited in textbooks of endocrinology as a basic mechanism for the development of functional and structural abnormalities in eyes, nerves, and kidneys as well as large arteries. However, generally no explanations are provided to explain exactly why the beta cell itself, whose functional failure is responsible for both the initial hyperglycemia and the adverse effects of chronic hyperglycemia on other tissues, also undergoes relentless loss of function over the course of the disease. This leads to my proposition of a vicious cycle in which the initial hyperglycemia that causes loss of normal function in other tissues might also cause continuing damage in beta cells, which in turn worsens the degree of hyperglycemia and worsens complications in other body tissues. This leads to a case of double jeopardy in which the initial abnormal function of the beta cell that causes hyperglycemia over time further damages the beta cell itself, causing it to undergo progressive decline in function. In other words, the beta cell becomes a secondary complication of the hyperglycemia it causes.
The harmful effects of chronic hyperglycemia are generally referred to as glucose toxicity. In regard to the beta cell, in the 1990s it was demonstrated in beta cell lines that prolonged culturing in media containing high glucose concentrations caused the loss of insulin secretion, which was preceded by loss of the critical transcription factors PDX-1 and MafA [1-3]. Up to a point, these losses were reversible if the elevated glucose concentrations in the culture media were removed and replaced by normal glucose concentrations [4]. This became a laboratory model for understanding the mechanisms of action of supraphysiologic glucose concentrations in causing loss of insulin gene transcription factors. Further studies demonstrated that the loss of Pdx-1 is post-transcriptional in nature, whereas the loss of MafA is post-translational [5].
Reactive oxygen species (ROS) are normally formed during metabolism of glucose and in normal concentrations participate in many physiologic processes, including regulation of gene expression, protective effects against bacterial invasion, cell growth, differentiation, apoptosis, autophagy, and senescence [6]. However, in the presence of prolonged elevated glucose concentrations, excessive formation of intracellular ROS occurs in several pathways of glucose metabolism. In physiologic levels glucose is normally metabolized via oxidative phorphorylation, which generates normal concentrations of ROS. However, when glucose concentrations are abnormally high over long periods of time, the oxidative phosphorylation pathway becomes flooded with excess glucose. This causes diversion of glucose to other pathways, including methylglycoxal/glycation, enediol formation, DAG/PKC activation, hexosamine metabolism, and sorbitol metabolism [7] (Figure 1). All of these additional pathways also generate ROS. A connection between the toxic effects of high glucose concentrations on beta cell function was established in studies demonstrating that exposure of beta cells to exogenous anti-oxidants in vitro and in vivo protected them from glucose toxicity [8-10]. This led to the conclusion that oxidative stress is likely mechanism for the adverse effects of hyperglycemia on beta cell function.
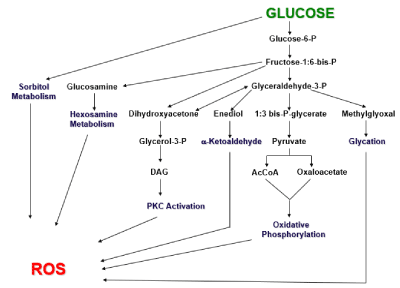
Figure 1. Origins of reactive oxygen species (ROS). Six biochemical pathways by which glucose metabolism can form reactive oxygen species (ROS) are shown. Under physiologic conditions, glucose primarily undergoes glycolysis and oxidative phosphorylation. Under the pathophysiologic condition of hyperglycemia, excessive glucose levels can swamp the glycolytic process and be shunted to the other pathways, with the outcome of forming excessive ROS levels. (Taken from reference 7)
A further complication to a scenario in which ROS generated by metabolic tissues during prolonged exposure to high glucose levels is the fact that the beta cell is unique in regard to the extent it is particularly prone to the damaging effects of oxidative stress. Compared to other tissues, the beta cell is poorly endowed with endogenous antioxidants. Glutathione peroxidase, in particular, is barely discernible [11-13]. This state of affairs leaves the beta cell especially at risk for damage by lipid peroxides, which are normally catabolized by glutathione peroxidase. Although superoxide dismutase is found in beta cells, it metabolizes superoxide and produces hydrogen peroxide, another potentially harmful ROS.
Clinical studies have suggested that subjects with type 2 diabetes can partially recover beta cell function if their hyperglycemia is reversed by short treatments of insulin [14,15]. This is an important observation because it argues for immediate therapeutic control of hyperglycemia in people who develop even minimal levels of hyperglycemia. This argument is not universally subscribed to in general practice. Rather, a “wait and see” approach is often taken to ascertain whether patients will truly develop frank diabetes. Resolution of these different points of view would require controlled studies comparing the beneficial effects of early treatment with and without pharmacologic intervention with careful matching for the effects of weight loss and exercise. Important phenomena such as consequences on beta cell apoptosis and beta cell replication would be key information although these measures cannot be documented clinically. However, this information can be readily obtained through animal studies. We reported such a study in which male ZDF rats were fed a high fat diet to induce hyperglycemia [16]. These animals have a genetic abnormality similar to type 2 diabetes that is not evident when they are fed a regular diet, but very quickly becomes manifest when fed a high fat diet. The animals were divided into two groups that were treated identically except one group received regular diets and the other received high fat diets for increasing durations of time. They were fed the high fat diets for up to 28 days and then returned to regular diets for 3 weeks. They were then sacrificed for procurement of pancreatic tissue for analysis. The purpose of the return to regular diets before sacrifice was to allow time for recovery from any damage caused by the high fat diet. The principal findings of this study were that by 9 days of exposure to hyperglycemia caused by the high fat diet, there was evidence for beta cell structural abnormalities and increased markers of ROS, After the three-week recovery period during which regular diets were fed, normoglycemia was reestablished and the structural abnormalities disappeared. Importantly, there were no increases in apoptosis or beta cell proliferation, which implies the normalization of beta cell structure and function were due to self-repair of existing beta cells. These favorable outcomes in the early stages of the experiments were accompanied by activation of intra-nuclear Nrf2, the principal regulator of endogenous anti-oxidant gene expression, as well as increases in expression of endogenous anti-oxidant hemoxygenase-2 (Figure 2). These findings provided evidence that in the early stages of beta cell damage caused by hyperglycemia-induced oxidative stress, endogenous anti-oxidant gene expression was activated, which led to self-repair of beta cells.
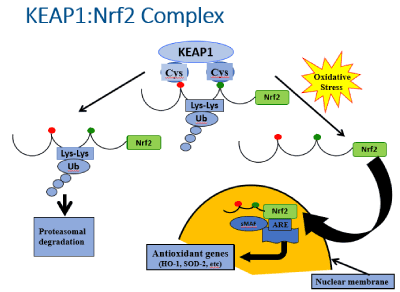
Figure 2. The KEAP1:Nrf2 complex. Nrf2 (nuclear factor erythroid-derived-2-related factor) is generally considered to be the master regulator of antioxidant gene transcription. Under quiescent conditions, Nrf2 is bound by KEAP1 (Kelch-like ECH-associated protein 1) in the cytoplasm, which ushers Nrf2 to proteosomes for degradation. However, in the face of oxidative stress, Nrf2 is released from KEAP1 and it enters the nucleus where it serves as a key activator of the promoter for antioxidant genes. (Taken from reference 16)
It is well documented that tissue repair occurs in retinae, kidneys, nerves, and arteries in diabetic patients who undergo successful pancreas transplantation and re-establishment of normoglycemia. It seems possible that early treatment of mild hyperglycemia of patients with type 2 diabetes may facilitate a degree of beta cell repair with the possibility of a return towards normal function and decreased progression of their disease.
- Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF (1992) Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest 90: 320-325. [Crossref]
- Olson LK, Sharma A, Peshavaria M, Wright CV, Towle HC, et al. (1995) Reduction of insulin gene transcription in HIT-T15 β-cells chronically exposed to a supraphysiologic glucose concentration is associated with loss of STF-1 transcription factor expression. Proc Natl Acad Sci U S A 92: 9127-9131. [Crossref]
- Poitout V, Olson LK, Robertson RP (1996) Chronic exposure of βTC-6 cells to supraphysiologic concentrations of glucose decreases binding of the RIPE3b1 insulin gene transcription activator. J Clin Invest 97: 1041-1046. [Crossref]
- Gleason CE, Gonzalez M, Harmon JS, Robertson RP (2000) Determinants of glucose toxicity and its reversibility in the pancreatic islet ß-cell Line, HIT-T15 cells. Am J Physiol Endocrinol Metab 279: E997-E1002.
- Harmon JS, Stein R, Robertson RP (2005) Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem 280: 11107-11113.
- Hopkins RZ, Li YR (2017) Essential of free radical biology and medicine. Cell Med Press.
- Robertson RP (2004) Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem 279: 42351-42354.
- Tanaka Y, Gleason CE, Tran POT, Harmon JS, Robertson RP (1999) Prevention of Glucose toxicity in HIT-T15 cells and Zucker diabetic fatty (ZDF) rats by antioxidants. Proc Natl Acad Sci USA 96: 10857-10862. [Crossref]
- Tanaka Y, Tran PO, Harmon J, Robertson RP (2002) A role for glutathione peroxidase in protecting pancreatic B cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci USA 99: 12363-12368.
- Mahadevan J, Parazzoli S, Oseid E, Hertzel AV, Bernlohr DA, et al. (2013) Ebselen treatment prevents islet apoptosis, maintains intranuclearPdx-1 and MafA levels, and preserves β-cell mass and function in ZDF rats. Diabetes 62: 3582-3588.
- Grankvist K, Marklund S, Taljedal IB (1981) Superoxide dismutase is a prophylactic against alloxan diabetes. Nature 294: 158-160.
- Tiedge M, Lortz S, Munday R, Lenzen S (1998) Complementary action of antioxidant enzymes in the protection of bioengineered insulin-producing rinm5f cells against the toxicity of reactive oxygen species. Diabetes 47: 1578-1585.
- Tonooka N, Oseid E, Zhou H, Harmon J, Robertson RP (2007) Glutathione peroxidase protein expression and activity in human islets isolated for transplantation. Clin Transplant 21: 767-772.
- Vague P, Moulin JP (1982) The defective glucose sensitivity of the B-cell in non-insulin-dependent diabetes: improvement after twenty hours of normoglycaemia. Metabolism 31: 1139-1142. [Crossref]
- Garvey WT, Olefsky JM, Griffin RF (1985) The effect of insulin treatment on insulin secretion and insulin action in Type II diabetes mellitus. Diabetes 34: 222-234.
- Abebe T, Mahadevan J, Bogachus L, Hahn S, Black M, et al. (2017) Nrf2/antioxidant pathway mediates β cell self-repair after damage by high-fat diet-induced oxidative stress. JCI Insight 2: 92854. [Crossref]