The current COVID-19 pandemic has presented unprecedented challenges to the world community. No effective therapies or vaccines have yet been established. Upon the basis of homologies to similar coronaviruses, several potential drug targets have been identified and are the focus of both laboratory and clinical investigation. The rationale for several of these drug candidates is presented in this review. Emerging clinical data has revealed that severe COVID-19 disease is associated with heightened inflammatory responses and a procoagulant state, suggesting that patient treatment strategies must extend beyond antiviral agents. Effective approaches to the treatment of vulnerable patients with comorbidities will render COVID-19 substantially more manageable.
COVID-19, SARS-CoV-2, coronavirus, antiviral, inflammation, coagulation
In December 2019, a novel virus was reported in Wuhan, China which caused severe respiratory distress and death in some patients [1,2]. The 2019 virus was identified as a novel coronavirus (CoV) named SARS-CoV-2 which causes COVID-19. By mid-March 2020, a world-wide pandemic was recognized with tens of thousands of people infected, and thousands of deaths reported. At this time, neither vaccines nor FDA-approved drugs are available for the treatment of COVID-19 patients. Governments world-wide have instituted travel restrictions, quarantines, and limitations on business activities and public events. The public is responding with enhanced hygiene and social distancing. These efforts will hopefully slow the rate of the rise in numbers of infected individuals and those requiring critical care. The social and economic crises resulting from this pandemic are unprecedented, and the cost will be measured in the trillions of dollars.
This virus can be defeated. Scientific efforts are in progress worldwide for the development of vaccines and drugs. A substantial body of scientific information currently exists on the biological and biochemical properties of similar viruses. Several potential drug candidates are currently being investigated. Clinical trials have been initiated in China, the United States and elsewhere and anecdotal reports present some positive signs.
Many of the drugs currently under consideration include those with FDA approval, but for diseases other than COVID-19. The pharmacology and potential side effects of these drugs in humans are known, and re-tasking these drugs to treat COVID-19 patients could be expedited. Additional FDA nonapproved drugs have been developed for similar viruses and tested in animals or in in vitro models, and these drugs will be advanced through the pipeline. SARS-CoV-2 is under intense scrutiny in several laboratories, and all vulnerabilities of the virus will be the focus of biochemical and structural studies. Assuming adequate funding to enhance the research infrastructure, targeted therapies will be developed. The purpose of this review is to outline a portion of the existing body of scientific information which ultimately might form the basis for a cure.
Coronaviruses (CoVs) with low pathogenicity infect the upper respiratory tract and cause mild cold-like symptoms. The CoVs can also cause intestinal tract infections. The highly pathogenic CoVs, including SARS-CoV (severe acute respiratory syndrome) and MERS-CoV (Middle East respiratory syndrome) cause predominantly lower airway disease, potentially including fatal pneumonia. The severe pneumonia is characterized by rapid virus replication, elevated cytokine and chemokine responses, infiltration of inflammatory cells and acute lung injury.
Factors influencing disease severity include initial infectious dose, age and comorbid conditions including diabetes, obesity, heart failure, renal failure and immunosuppression. The clinical presentation can occur in three phases [1,2]. In the initial phase, there is rapid viral replication within the lungs, accompanied by fever, cough and other symptoms which can resolve within days. In the second phase, high fever and hypoxemia can progress to pneumonia symptoms. Approximately 20% of COVID-19 patients progress to the third phase which includes acute respiratory distress syndrome (ARDS) and frequently death [1,2]. In SARS patients with severe disease, high serum levels of pro-inflammatory cytokines (IFN-g, IL-1, IL-6, IL-12 and TGFβ and chemokines (CCL2, CXCL10, CXCL9 and IL-8) accompanied by low levels of the anti-inflammatory cytokine IL-10 are found [3]. In human and animal studies, monocyte-macrophages and neutrophils accumulate in the lungs and these cells can be a source of inflammatory cytokines and chemokines.
One of the first clusters of COVID-19 patients was treated at Wuhan Jinyintan Hospital, China in January 2020 [1]. Of the 99 patients, most were older (average age 55.5), and more were male (67 men, 32 women). Most patients presented with fever and cough. Some also presented with shortness of breath, sore throat, muscle ache, confusion, headache, and gastrointestinal distress. Imaging studies revealed most patients had bilateral pneumonia. Some developed ARDS (17%) and of these, several patients (11%) worsened and ultimately died of multiple organ failure.
About half of the treated patients had underlying chronic diseases including cardiovascular and cerebrovascular disease and diabetes. Among the first patients to die, the first two had a long history of cigarette smoking. Many of the severely ill patients had bacterial or fungal coinfections.
All patients in the Wuhan studies were treated in isolation, some with mechanical ventilation. Patients were provided standard antimicrobial therapy, but the impact of these medications on patient outcome was not studied systematically. Most received antiviral therapy consisting of oseltamivir, ganciclovir, lopinavir and ritonavir for 3 to 14 days. Most were also given antibiotics including cephalosporins, quinolones, carbapenems, tigecycline, linezolid and antifungal drugs. Some (19%) were also treated with methylprednisolone and dexamethasone.
A second cohort of 138 COVID-19 patients was treated at the Zhongnan Hospital in Wuhan also in January of 2020 [2]. In this study, the average age of the treated patients was 56 years, and 54.3% were men. As with the above group, 16% developed ARDS, but fewer died (4.3%). Patients were treated with oseltamivir, moxifloxacin, ceftriaxone, azithromycin and glucocorticoids. The median neutrophil count in patients who were treated in the ICU (4.6 × 109 /L) was significantly higher (p<0.001) than that of the non-ICU patients (2.7 × 109 /L) which might suggest neutrophil count as a marker of potential disease progression.
The profiles of the patients in these two groups are similar, and fit the pattern identified in previous studies of MERS-CoV and SARS-CoV. The subset of patients who progressed to respiratory distress likely suffered a “cytokine storm” which is fatal in a substantial number of patients. The potential benefits of the antimicrobial therapy which differed between the groups is as yet unknown. Due to the apparent three phases of the disease, the timing of therapy will be a critical factor.
Data obtained from those hospitalized with COVID-19 has yielded important observations regarding frequent laboratory abnormalities during infection. A systematic analysis of 43 separate studies, which combined data from 3,600 patients, revealed elevated C-reactive protein (CRP) in 69%, lymphopenia in 57%, and increased lactate dehydrogenase (LDH) in 52% [4]. Alteration of some clinical biomarkers may signal impending clinical decline or poor prognosis. It has been noted that lymphopenia and an increase in the neutrophil to lymphocyte ratio is associated with an increase in hospital mortality [5]. Both CD4+ and CD8+ lymphocyte subsets are decreased during severe infection [6]. CRP has also been suggested to help predict severity. In an analysis of 27 COVID-19 patients, receiver-operator characteristic analysis demonstrated that at a cut off of 20.4 mg/L, the CRP had an 83% sensitivity and 91% specificity for predicting severe disease [7]. A useful scoring model (termed “CALL” by investigators) uses the LDH and lymphocyte count in combination with patient age and comorbidities to estimate the 5- and 10-day risk of disease progression [8].
SARS-CoV-2 RNA
SARS-CoV-2 is a coronavirus (CoV) and member of the Coronaviridae family and Orthocoronavirinae subfamily which includes the α, β, g and d-coronaviruses [9]. The β-coronavirus subfamily includes SARS-CoV which causes the severe acute respiratory syndrome (SARS), SARS-CoV-2, which causes COVID-19, and MERS-CoV, which causes Middle East respiratory syndrome (MERS).
Coronaviruses primarily cause infections in birds and some mammals, but occasionally can cross species barriers and infect humans as well. In December 2019, a novel coronavirus was identified in a cluster of patients with pneumonia in Wuhan, China. This novel coronavirus, SARS-CoV-2, was subsequently named 2019-nCoV by WHO. An alarming and unusual characteristic of this virus is that it spreads from human to human.
The viral genome of SARS-CoV-2 is an enveloped, single-stranded positive-sense RNA approximately 30 kb in length and it shares high sequence homology with SARS-CoV [10]. Upon the basis of its genome sequence, sensitive and specific clinical assays could be developed using real time-RT-PCR to measure viral RNA.
SARS-CoV-2 proteins
Coronaviruses are large, about 100-120 nm in diameter (Figure 1). The spike proteins protruding from the envelope of the coronavirus resemble a crown accounting for the name. The β-coronaviruses SARS-CoV and MERS-CoV share high sequence identity and similarity with SARS-CoV-2, and the overall gene organization is similar. All have a surface Spike glycoprotein (S) small envelope (E), matrix protein (M) and nucleocapsid protein (N). SARS-CoV-2 also has sixteen non-structural (nsp) accessory proteins including the leader protein (nsp1), the papain-like protease (PL-pro, nsp3), the 3-chymotrypsin, 3C-like protease (3CL-Pro, nsp5), the RNA-directed RNA polymerase (RdRp, nsp12), the helicase (nsp13), the guanine N7-methyltransferase (nsp14) the uridylate-specific endoribonuclease (nsp15), the 2’-O-methyltransferase (nsp16) and the ORFa protein [10]. Several of these proteins currently are targets for inhibitory drugs as described below, and all are potential targets.
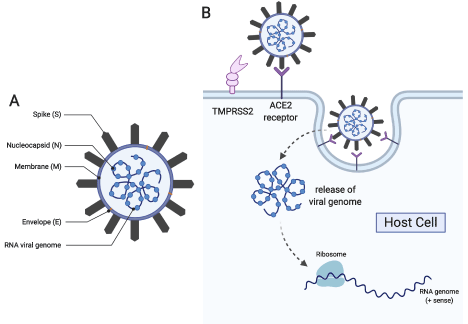
Figure 1. (A) Schematic of the COVID-19 virus showing critical viral proteins and (B) its route towards host cell infection and propagation (This figure was generated with biorender.com)
SARS-CoV-2 life cycle
Understanding the events in the lifecycle of SARS-CoV-2 is essential to identify mechanism-based targets which can interfere with infection and propagation of the virus. The life cycle of SARS-CoV-2 shares many common steps with other coronaviruses [11,12]. In the first step, the S-protein of the virus binds to hosts cells with the ACE2 protein on the host cell surface [13,14]. Proteases on the host-cell membrane, such as the serine protease TMPRSS2, modify the viral spike protein. If this step is blocked, the virus cannot enter the cell. After host protease modification, the envelope of the virus fuses with the host cell membrane initiating endocytosis. Once the enveloped virus has entered the cell, an uncoating process occurs and the viral RNA is released into the host cell. Using host translational machinery, the viral RNA is translated into a large polyprotein containing the sequences of multiple proteins. The polyprotein is then cleaved by viral proteases into individual, functional peptides. The virally encoded RNA polymerase then makes multiple copies of the viral RNA. Viral RNA is assembled with the nucleocapsid N and other viral proteins, and host cell membrane components provide the envelope of the new virus particles. The next, obligate step in the life cycle is the release of the newly formed virus particles by cleavage of glycoprotein bonds.
Pharmacological considerations for the inhibition of SARS-CoV-2
Systems to study anti-SARS-CoV-2 pharmacology: The development of antiviral drugs generally begins with in vitro studies. Initial studies of in vitro SARS-CoV-2 utilized an isolate from a January, 2020 Washington State, U.S. patient after four passages in Vero (African green monkey) E6 cells [15]. Replication was examined several human cell lines including adenocarcinoma (A549), liver (HUH7.0), and embryonic kidney cells (HEK-293T), in addition to African green monkey kidney (Vero) E6 and Vero CCL81 cells. Cytopathic effects (CPE) were only observed in Vero cells, with replication titers >107 plaque forming units (PFU) at 24 hours post infection, and slightly higher in Vero E6 cells. HUH7.0 and 293T cells showed only modest viral replication, and there was no evidence of SARS-CoV-2 replication in A549 cells [15].
For evaluation of antiviral drug activity, several strategies are employed. In silico screening has been used when the target of potential drugs is a viral protein of known atomic structure, such as viral polymerases or proteases essential for replication. Experimentally, biochemical assays for the activity of such viral enzymes has also been used to efficiently screen large libraries of compounds. However, initial efforts for emergency situations such as COVID-19 must also focus on repurposing drugs already used in clinical trials or licensed for other viral infections or diseases. In this case, assays generally involve testing the ability of drugs to either inhibit CPE in cell cultures, an indirect measure of antiviral activity, or measuring directly the inhibition of replication by assaying replication kinetics using RT-qPCR to measure viral RNA replication or plaque-forming units generated in treated versus sham (vehicle)-treated cells.
For both of these approaches, Vero E6 cells are ideal because they support robust SARS-CoV-2 replication and undergo severe CPE [15]. Although human cells would be ideal to perfectly match the cellular factors essential for viral replication, African green monkeys are very close relatives. Their version of the Angiotensin converting enzyme 2 (ACE2) cellular receptor for SARS-CoV-2 is presumably nearly identical to that of the human homolog [16].
The approaches for antiviral evaluation generally include beginning with pre-treatment of cells with a drug of interest, followed by infection and then continued treatment to maximize the sensitivity to measure an antiviral effect. Ideally, drugs should show some initial antiviral effect, usually measured in log10 orders of magnitude in viral titer reduction at doses that are not toxic to the cells. Drugs that also inhibit infection when applied only after infection of the cells, more realistically reflecting treatment of a recently diagnosed or exposed patient, are especially promising. Effective antiviral doses should of course result in physiologic levels with no known toxicity in human clinical trials. Viral doses used in antiviral assays can also vary but are generally large enough to produce robust and consistent replication.
A variation on the typical antiviral assay described above is to genetically engineer the virus of interest to express a reporter gene such as green fluorescent protein or luciferase during replication. This enables the readout of antiviral replication to be a simple, colorimetric assay instead of the more complex assays of viral RNA (RT-qPCR or plaque assay). Such viruses have recently been described for SARS-CoV-2 [17].
Assays such as those described above with SARS-CoV-2 are currently recommended to be conducted in biosafety level 3 containment because they rely on wild-type virus. The development of attenuated virus strains will eventually reduce the containment requirement for these antiviral assays. Another approach is the development of viral replicons, truncated viral genomes that can replicate in cells after introduction of the RNA into the cytoplasm; this can be accomplished packaging the replicons with viral proteins expressed from a different cDNA construct, and the resulting replication cannot produce infectious virus due to the absence of an essential viral gene. Replicons can also be combined with the expression of a reporter gene such as GFP or luciferase to simplify the readout of antiviral activity (reduction in reporter activity) combined with the lower biocontainment needs.
Drugs can potentially interfere with binding and entry of viruses into human cells: Upon the basis of the viral life cycle discussed above, several potential drug targets can be identified. The spike protein of SARS-CoV-2 utilizes the ACE2 protein present on the surface of host lung cells as a docking site. The spike protein would therefore likely be a good target for vaccine development. Patients who survived SARS infection have neutralizing antibody toward the SARS S protein [13]. However, the spike protein must be modified by host cell proteases prior to viral entry. The TMPRSS2 serine protease has been identified as the likely protease. Two drug candidates, camostat and nafamostat (Figure 2) are known inhibitors of TMPRSS2. In vitro studies show that nafamostat is a potent inhibitor of the serine protease which facilitates MERS-CoV infection [18]. In SARS-CoV-2 infected Vero E6 cells, the half-maximal effective concentration (EC50) of nafamostat was shown to be 22.50 µM [19]. Camostat inhibits the serine protease important for influenza viral entry [20] and it is approved in Japan for the treatment of chronic pancreatitis [13].
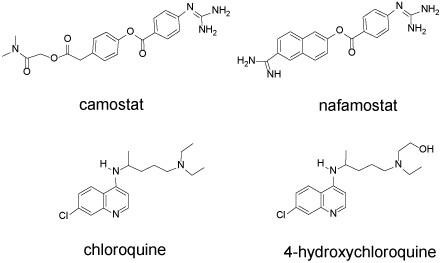
Figure 2. Camostat, nafamostat, chloroquine, hydroxychloroquine. These drugs interfere with the initial stages of SARS-CoV-2 infection
Following engagement of the ACE2 receptor and protease cleavage of a portion of the spike protein, the envelope of the virus begins to merge with the host cell membrane, initiating endocytosis. Further processing of the viral proteins must occur within the endocytic vesicle, and such processing requires an acidic pH. Chloroquine (CQ) and hydroxychloroquine (HCQ) are 4-aminoquinolones (Figure 2) that concentrate within the vesicles and increase the pH due to their basic functional groups.
In Vero E6 cells, HCQ (EC50=0.72 µM) is more potent than CQ (5.47 µM) in inhibiting SARS-CoV-2 [21]. Wang and coworkers reported a lower EC50 value for chloroquine (1.13 µM) [19]. In a recent clinical study of 36 French COVID-19 patients (RT-PCR positive), the effect of HCQ on reducing SARS-CoV-2 RNA was measured. HCQ (200 mg × 3 times per day × 10 days) was provided to 20 patients while 16 served as untreated controls. Among the HCQ treated group at day 6, only 40% remained PCR-positive while 90% of the control group remained positive. Six patients received simultaneous azithromycin (500 mg day 1, 250 mg/day 4 days) none of whom remained virus positive at day 6. No other clinical observations were reported [22].
Chloroquine is a widely used anti-malarial drug and is also used to treat autoimmune disease. The chloroquines change the number, size and morphology of endosomes and endolysosomes, explaining in part their anti-malarial activity [23]. Acidification is important for endosome maturation and function. Drug induced elevation of pH blocks the intracellular transport of the virus and release of new virus particles. In glioblastoma cells, HCQ inhibits acidification of lysosomes blocking autophagy [24]. The mechanism for the anti-inflammatory activity of CQ and HCQ is not well understood. HCQ and CQ are substrates for P450 enzymes and therefore can alter the pharmacology of other drugs modified by P450. CQ use can increase plasma concentrations of digoxin, and HCQ use can increase levels of metoprolol [23].
Chloroquine and hydroxychloroquine are not devoid of toxicity and both have a narrow therapeutic window. Patients with autoimmune disease treated with standard concentrations of CQ or HCQ for years have increased risk of developing macular retinopathy. The serum concentration of CQ and HQ can take several weeks to build up to therapeutic levels [25]. However, a bolus dose of either can be acutely toxic and result in cardiac arrest. The normal dose of HCQ is 500-600 mg/day. The lethal dose is not well established; however, a fatal outcome is associated with ingestion of greater than 5 g. Chloroquine is 2-3 times more toxic than HQ and a lethal dose is 30 mg/kg. Chloroquine intoxicated patients present with tachycardia and hypotension and a widening QRS interval. Diazepam is recommended for seizures and dysrhythmias in intoxicated patients and alkalinization with sodium bicarbonate is used for QRS widening and hypotension [26].
Drugs can interfere with the translation and proteolysis of the polyprotein complex: Nitazoxanide (NTZ) is an FDA approved antiprotozoal agent is (Alinia, 500 mg/day) used primarily for intestinal infections. It has been repurposed for the treatment of influenza [27]. Nitanoxanide is hydrolysed intracellularly to tianoxanide (Figure 3). NTZ has multiple modes of action in different virally infected cells [28]. In HCV infected cells, nitazoxanide induces the phosphorylation of the dsRNA-dependent protein kinase (PKR) which then phosphorylates the eukaryotic transcription initiation factor 2 alpha (eIF2a) as part of an innate immune response which results in the inhibition of translation of the viral RNA [29,30]. In cells infected with influenza, NTZ blocks post-translational maturation of viral hemagglutinin [27]. In MERS-infected cells, NTZ inhibits processing of the viral N protein, needed for packaging RNA into the virus capsule [31]. In a screen for anti-coronavirus drugs, NTZ was identified as an agent that inhibited expression of the nucleocapsid N-protein, needed for the packaging of the viral RNA [32]. In COVID-19 infected Vero cells, NTZ was a potent inhibitor of virus replication with an EC50 of 2.12 µM [19].
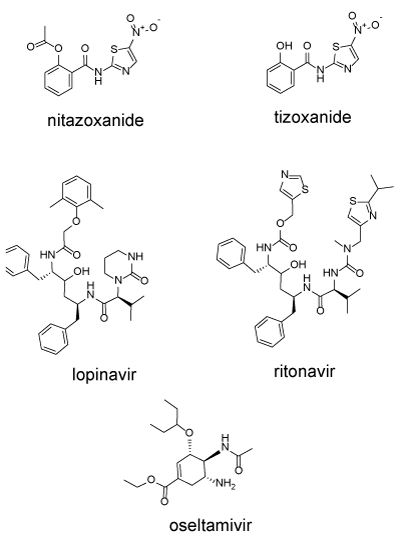
Figure 3. Nitanoxanide, tizoxanide, lopinavir, ritonavir, oseltamivir. These drugs can interfere with multiple stages of viral infection
Cleavage of the polyprotein into the component proteins is accomplished by virally encoded proteinases including the papain-like protease (PL-pro, nsp3), the 3-chymotrypsin, 3C-like protease (3CL-Pro, MPro or main proteinase, nsp5). Two protease inhibitors were designed to inhibit HIV-encoded proteases lopinavir and ritonavir (Figure 3). Both drugs are usually administered as a combination (Kaletra). Ritonavir is added as a P450 inhibitor to reduce metabolism of and boost concentrations of lopinavir. This combination has been shown to inhibit SARS and was used in one of the Wuhan studies [1].
In a recent randomized clinical trial of 199 patients with confirmed SARS-CoV-2 infection, the combination lopinavir (400 mg) and ritonavir (100 mg) did not improve time to improvement or mortality. Gastrointestinal adverse events were more common in the lopinavir-ritonavir group [33]. Li and DeClercq have questioned whether lopinavir and ritonavir, designed specifically as HIV protease inhibitors, would be appropriate inhibitors for the SARS-CoV-2 main protease [34]. More targeted inhibitors are undoubtedly in the pipeline.
Drugs can interfere with the replication of viral RNA: A single viral RNA molecule injected into a human cell can be replicated by the RNA-directed RNA polymerase (RdRp, nsp12), generating multiple copies. Nucleoside analogs represent the earliest class of antiviral drugs. Ellion et al. first developed acyclovir for the treatment of herpes simplex infections in the 1970’s [35]. Development of similar analogs led to successful treatments for cytomegalovirus in immunocompromised cancer patients. Nucleoside analogs including azidothymidine (AZT) dideoxycytidine (ddC) were then developed for the treatment of human immunodeficiency virus, HIV [36].
All nucleosides analogs undergo metabolic activation in infected cells to the corresponding 5’-triphosphates (NTP). In order to have antiviral activity, the NTP must be incorporated into the growing RNA chain, and once incorporated they act as chain-terminating substrates, induce lethal mutagenesis, or otherwise interfere with RNA function. Agents examined in vitro in Vero E6 cells infected with SARS-CoV-2 include the following drugs and their corresponding EC50: favipiravir (61.88 µM), ribavirin (109.50 µM), penciclovir (95.96 µM) and remdesivir (0.77 µM) (Figure 4).
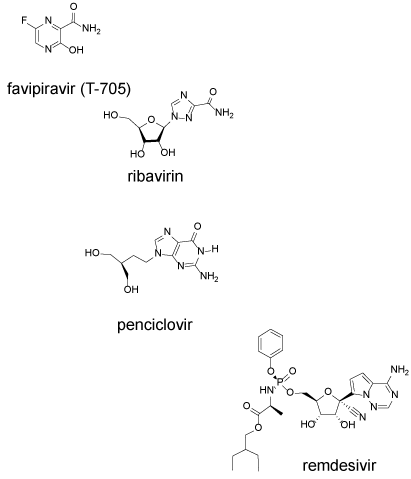
Figure 4. RNA polymerase inhibitors favipiravir, ribavirin, penciclovir and remdesivir inhibit replication of the viral RNA
Favipiravir (6-fluoro-3-hydroxy-2-pyrazinecarboxamide) is a broad-spectrum antiviral agent and it is chemically the simplest of the group. Favipiravir has received FDA approval for treating influenza. Upon entry into a cell, it is converted to the nucleoside analog by the purine salvage pathway enzyme, hypoxanthine-guanine phosphoribosyl transferase (HGPRT) [37]. The nucleoside is then converted to the mono, di and triphosphate analog which is a substrate for the RNA polymerase. In vitro studies suggest that it can both act as a chain terminator and induce miscoding and lethal mutagenesis [38] (Table 1). An additional mechanism of action could be related to the instability of the nucleoside analog which hydrolyzes in water with a half-life of 15 h at neutral pH [39]. Hydrolysis after incorporation could similarly inactivate the RNA chain. At higher concentration, favipiravir inhibits RNA elongation, however, at lower concentration noninfectious virus particles are produced [40].
Table 1. Drugs tested in Vero E6 cells infected with SARS-CoV-2. EC50 values µM
Drug |
EC50 (μM) |
Status |
Reference(s) |
Remdesivir |
0.77 |
Not FDA approved
In clinical trials |
19 |
Hydroxychloroquine |
0.72 |
FDA approved for malaria, rheumatoid arthritis, lupus erythematosus |
21 |
Chloroquine |
1.13, 5.47 |
Approved for malaria and amebiasis |
19,21 |
Nitazoxanide |
2.12 |
FDA approved for diarrhea caused by Giardia lambia |
19 |
Nafamostat |
22.5 |
Not FDA approved
In clinical trial |
19 |
Favipiravir |
61.88 |
Not FDA approved
In clinical trial |
19 |
Penciclovir |
95.96 |
FDA approved for topical treatment of cold sores |
19 |
Ribavirin |
109.50 |
FDA approved for chronic hepatitis C infection |
19 |
Ribavirin is a triazole-3-caboxamide ring connected to a ribofuranosyl sugar. Ribavirin is FDA approved for the treatment of chronic hepatitis C infection. Unlike favipiravir, ribavirin can be directly converted to the corresponding phosphates by host kinases. Ribavirin-triphosphate (RTP) can then be then incorporated into the growing RNA chain, interfering with chain elongation. In influenza infected cells, ribavirin inhibits the host enzyme, inosine monophosphate dehydrogenase (IMPDH), the rate limiting enzyme for de novo purine synthesis. If the primary mechanism of ribavirin is inhibition of GTP synthesis, and favipiravir competes with GTP for incorporation into RNA, the combination of ribavirin and favipiravir might be synergistic.
Pencyclovir is a guanine analog, similar in structure to acyclovir, the first established antiviral drug for the treatment of herpes simplex infections, and ganciclovir, the acyclovir analog used to treat cytomegalovirus infection [36]. Pencyclovier is FDA approved for the topic treatment of cold sores. Pencyclovir, acyclovir, ganciclovir are all nucleoside analogs that must be converted intracellularly to the corresponding 5’-triphosphates (NTP). As the NTP, the analogs can be substrates for incorporation by RNA or DNA polymerases. All members of this family have an intact 5’-hydroxyl for conversion to the NTP, but all have defective 3’-hydroxyls which are either absent or structurally altered, preventing chain elongation.
Remdesivir is the most chemically complicated and difficult to synthesize of the nucleoside analogs [41]. Remdesivir is in clinical trial as an HIV reverse transcriptase inhibitor. Remdesivir is a phosphoramidate prodrug resembling adenosine 5’-monophosphate. The rate limiting step in the biological activation of the nucleoside analogs such as penciclovir is the intracellular enzymatic conversion to the nucleotide. Nucleotides do not easily cross the cell membrane, and so the 5’-phosphate of remdesivir is protected as a phosphoramidate which allows entry into the cell. Once in the cell, the protecting groups are hydrolyzed generating the charged 5’-monophosphate. As with other drugs in this class, remdesivir is then further converted to the 5’-triphosphate which interferes with RNA replication.
A 34-year-old male COVID-19 patient in the United States was treated with intravenous remdesivir after 7-day hospitalization with persistent fever and the development of radiographic pneumonia. The patient’s clinical condition improved the following day [42]. Furthermore, observational data from a cohort of 53 patients receiving remdesivir through a compassionate use program reported improvement in 68%. Despite this cohort being quite ill (64% were receiving mechanical ventilation when remdesivir was initiated), the case fatality rate was 13%. When juxtaposed to mortality rates of 17-78% in severe cases in China, the use of this drug appears promising [43]. Randomized controlled trials are currently underway to definitively assess the safety and effectiveness of remdesivir.
Drugs can inhibit viral release
Olsetamivir (OS) (Figure 3) is a neuraminidase inhibitor. Neuraminidase is a viral protein anchored in the viral envelope. The major function of neuraminidase is to cleave the chemical link between sialic acids and adjacent sugar residues. If the neuraminidase activity is blocked, the newly synthesized virus accumulates on the host cell surface and cannot spread to other cells [44]. Oseltamivir (Tamiflu) is approved by the FDA for the treatment of influenza infection. The combination of oseltamivir plus azithromycin was found to be more effective than oseltamivir alone in a cohort of high-risk influenza patients [45]. Oseltamivir was among the drugs used in both Wuhan studies discussed above. Efficacy for COVID-19 is as yet unknown.
Pharmacological considerations for COVID-19 patients not targeted against SAS-CoV-2: Most patients infected with SARS-CoV-2 have mild symptoms and good prognosis. On the other hand, a subset of patients develops severe disease and some ultimately die. Currently, very little is known about the pathological manifestations of COVID-19. Identifying the characteristics of patients at higher risk for developing severe disease, as well as understanding the pathophysiological basis of severe disease could considerably reduce mortality from the COVID-19 pandemic. While advanced age and comorbidities including hypertension, immunosuppression and other coexisting disease are associated with severe COVID-19 disease and death, the underlying mechanisms for these associations are not well understood. Emerging studies suggest an overly exuberant immune response to viral infection can contribute to severe disease. Effective COVID-19 therapy will include not only antiviral therapy, but therapy targeted at potentially maladaptive immune responses.
Treatment of severely ill COVID-19 patients will be a challenge. COVID-19 patients with comorbidities who develop more severe disease have frequently been managed with an array of agents prior to infection. Significant questions remain as to how prior therapy might alter susceptibility to SARS-Cov-2 infection, if prior therapies can be continued during COVID-19 therapy, and what drug-drug interactions might exist for commonly prescribed drugs and anti-SARS-CoV2 drugs. Substantial further study will be required to answer these questions. Below, we discuss several areas which must be explored.
ACE2, the renin-angiotensin axis and COVID-19. The receptor for SARS-CoV-2 is the cell surface protein ACE2, or angiotensin-converting enzyme 2. ACE2 is a component of the renin-angiotensin-aldosterone system (RAAS) involved in regulating extracellular fluid volume [46]. The RAAS is the focus of multiple hypertension studies and several antihypertensive drugs have been developed to modify components of the system. ACE2 was discovered in 2000 [47,48] and in 2003, it was identified as the receptor for the SARS coronavirus [49]. The intersection of ACE2 and COVID-19 has led to considerable speculation on how the use of antihypertensive and other drugs might influence susceptibility and response to COVID-19.
The RAAS is comprised of a series of enzymes that cleave peptides, and a series of receptors for those peptides. Angiotensinogen is a 16 amino acid peptide which is cleaved by renin to a 10 amino acid peptide, AngI. AngI can be cleaved by the angiotensin converting enzyme (ACE) to the 8-amino acid peptide, AngII. AngII the binds to the cell surface receptor AT1R. Alternatively, AngI can be cleaved by ACE2 to a 9 amino acid peptide (ANG 1-9), and AngII can be cleaved by ACE2 to a 7 amino acid peptide (Ang 1-7). Ang 1-7 can bind to the receptor MasR. Activation of the ACE pathway can lead to vasoconstriction and hypertension whereas activation of the ACE2 pathway opposes the ACE pathway, resulting in vasodilation and lower blood pressure. Antihypertensive drugs are available to inhibit the protease activity of ACE (ACEIs, e.g., lisinopril), or to block the binding of AngII to AT1R (ARBs, e.g., losartan, valsartan).
Diaz has proposed that the use of ACEIs or ARBs increase the number of ACE2 receptors in the cardiopulmonary circulation, potentially increasing the risk of severe disease outcome from COVID-19 infection [50]. Diaz explains that the ACE2 connection might explain why patients with hypertension are overly represented among COVID19 patients with severe disease. The literature basis for the Diaz proposition is a study by Ferrario in 2005 [51]. In that study a series of 36 normotensive rats were given no drug, lisinopril, losartan, or both at levels slightly higher than normally used in humans. ACE2 mRNA activity in cardiac membranes was measured after 12 days. Lisinopril increased cardiac ACE2 mRNA by a factor of 5, but not ACE2 activity whereas losartan increased both by a factor of approximately 3. The implication is that losartan increased the number of ACE2 receptors, increasing chance or severity of infection. The potential extrapolation of this study to humans has not been studied.
In animal models of ARDS, age related changes in the ratio of ACE to ACE2 are observed in the pulmonary RAAS [52] with a shift toward greater aggravating inflammation and increased lung damage with increased age, perhaps suggesting why age is a comorbidity for severe COVID-19 disease. In a prospective observational study in neonates, children and adults on ventilation for ARDS, examination of BAL fluid revealed increasing IL-6, IL-10 and neutrophil myeloperoxidase (MPO) with increasing age, but surprisingly no changes were seem in the relative activity of ACE and ACE2. Increased severity of COVID-19 in older adults is unlikely related to age-related changes in the RAAS.
In African Americans, RAAS inhibition and low renin states may be present given the higher prevalence of salt-sensitivity in this population [53]. Consumption of a high salt diet in a salt sensitive state is associated with reduced plasma renin activity and angiotensin concentration [54]. This state is accompanied by paradoxical elevation in renal angiotensin II and tissue angiotensin I receptor expression reflecting oxidative stress on cardiovascular tissues [55,56]. Angiotensin II stimulates expression of PAI-1 and with an increase in PAI-1 from endothelial cells [57]. PAI-1 is a strong inhibitor of fibrinolysis. This pathway may partially explain the thrombogenic effects seen in COVID-19 patients, especially among African Americans [58].
A recent clinical study at the Shenzhen Third People’s Hospital in China examined the use of antihypertensive drugs in COVID-19 patients [59]. Of the 417 hospitalized patients, 42 were on anti- hypertensive therapy. Of these, 17 were using ACE inhibitors (ACE-I) or angiotensin-II receptor blockers (ARBS). The remaining 25 were using calcium channel blockers, β-blockers or diuretics. Of the patients on ACE-I/ARB therapy 4 of 17 (23.5%) progressed to severe disease while 12/25 (48%) in the other group developed severe disease and one died. Upon the basis of these findings, the author suggest RAAS inhibition with ACE-I or ARBs can improve the clinical outcome of COVID-19 patients with hypertension.
Well-designed large-scale clinical studies to clarify the benefits versus risks that ACE-I and ARBS in those with COVID-19 are desired. Although a definitive verdict has yet to be elucidated, the impact of withdrawing these agents, in those taking them for a variety of indications, is clearer. As outlined by Vaduganathan and colleagues, the benefits of maintaining ACEi and ARBS likely outweigh the risks of abruptly withdrawing them [60]. For example, those with myocardial infarction, initiation of ACEi reduces mortality (ACE Inhibitor Myocardial Infarction Collaborative Group, 1998). In those with congestive heart failure, withdrawal of treatments can lead to worsening clinical status [61,62] and decreased left ventricular ejection faction [63]. Therefore, in the setting of potential COVID-19 associated cardiac injury, it seems prudent to continue these cardioprotective medications. Guidance supporting the continuation of ACEi and ARBS is supported by several medical societies including the American College of Physicians [60].
A subset of COVID-19 patients develops high fever and progress to pneumonia while a smaller subset progress to acute respiratory distress. An exaggerated immune response and “cytokine storm” appear to accompany or drive this progression to ARDS. The timing of anti-inflammatory therapy is critical. Dampening the immune response early in the infection could allow more extensive viral spread. On the other hand, once the patient is in respiratory distress, attenuating the inflammatory response may be required to prevent irreparable lung damage. In SARS and MERS patients, the use of corticosteroids has not been shown to provide substantial benefit, and other options are needed.
NSAIDS (Non-steroidal anti-inflammatory drugs, Figure 5) are frequently used in patients with the flu to reduce fever and pain. Ibuprofen is a commonly used NSAID. However, Qiao and others reported that ibuprofen treatment for 8 weeks in rats could increase ACE2 levels in cardiac tissues, as with losartan above [64]. While this finding has not been replicated in humans, it has led some to suggest ibuprofen might be contraindicated in COVID-19 and, acetaminophen (paracetamol, Tylenol) might be more appropriate [65]. On the other hand, in a bovine model of respiratory syncytial virus (RSV) ibuprofen decreased disease severity possibly by decreasing the prostanoid surge that follows RSV infection [66].
Figure 5. NSAID anti-inflammatory agents aspirin, ibuprofen, indomethacin, acetaminophen and the macrolide antibiotic azithromycin
Although clinical anecdotes during the early phases of the pandemic have led to potentially misguided statements regarding avoidance of cyclooxygenase inhibitors, there is no clinical evidence to support or refute their use in those with COVID-19 [67]. During mild disease, these agents may help to alleviate symptoms, while in those who are critically ill, the potential for acute kidney injury and gastrointestinal bleeding temper their use.
Indomethacin is another NSAID, available only by prescription. While ibuprofen and indomethacin have comparable efficacy for the short-term treatment of rheumatoid arthritis, indomethacin causes a greater incidence of gastric irritation. Indomethacin, but not aspirin, blocks SARS-CoV RNA synthesis in Vero cells (ID50=5 µM) and was more effective than ribavirin [68]. While the mechanism of this inhibition is not known, indomethacin is known to be a potent inhibitor of murine neutrophil peroxidase and so it may have a role in attenuating permanent lung damage [69].
Azithromycin (Figure 5) has both anti-bacterial and anti-inflammatory effects. Azithromycin causes the degranulation of neutrophils which could facilitate an initial antimicrobial effect. However, following neutrophil degranulation, serum concentration of chemokines and IL-6 declined [70]. Macrolide antibiotics improve pulmonary function and decrease exacerbation of frequency for persons with diffuse panbronchiolitis or cystic fibrosis [71].
Immunomodulatory effects include decreasing production of reactive oxygen species, inhibiting neutrophil activation accelerating neutrophil apoptosis. Azithromycin has been used in the treatment of cystic fibrosis. Erythromycin and clarithromycin are widely used in Japan to treat COPD [71]. Azithromycin acutely stimulates PMN degranulation and phagocytosis-associated oxidative burst in humans, followed by inhibition of PMN oxidative burst for up to 28 days after last dose. Acute PMN activation may facilitate the killing of microorganisms while the suppression of chronic inflammation may limit airway damage [72]. Indomethacin and azithromycin may have benefit for COVID-19 patients, apart from any antiviral activity.
Tocilizumab is a humanized monoclonal antibody to target the IL-6 receptor. As previously mentioned, the exaggerated immune response and cytokine storm that occurs with COVID-19 is associated with the development of ARDS. IL-6 is believed to be an important proinflammatory cytokine in the development of ARDS. Blockade of the IL-6 signal transduction pathway with tocilizumab has been proposed to decrease the progression to severe lung disease. A case report [73] and an observational study of a small cohort of COVID-19 patients treated with tocilizumab [74] demonstrated a decrease in CRP, but clinical outcomes were variable. Tocilizumab has been associated with gastrointestinal perforation and bacterial infections when used for the treatment of rheumatoid arthritis and giant cell arteritis [75]. In those with critical illness, it is unclear if the benefits of tocilizumab outweigh its potential risks. Data from clinical trials using tocilizumab are needed to detail its efficacy and safety in patients with COVID-19.
COVID-19 and the coagulation cascade
Substantial prior literature has examined mechanisms associated with viral activation of immune responses and coagulation. Viruses activate the coagulation cascade by inducing tissue factor (TF) expression and by disrupting the endothelium [76] Activation of coagulation is beneficial for limiting pathogen dissemination, destroying pathogens and facilitating tissue repair. However, overactivation can lead to thrombosis, excessive inflammation and tissue damage [77]. Following viral infection, proteolysis of fibrin generates the fibrin degradation products (FDP) E-fragment, D-monomer and D-dimer. The FDP are potent chemotactic proteins that induce neutrophil migration. While neutrophils can function to eliminate infectious organisms, they can also cause substantial oxidative host injury [78].
With respect to coronavirus infections, severe disease is driven by inflammatory cell recruitment and damage to pneumocytes, suggesting a connectivity between virus infection, pro-inflammatory cytokine responses and coagulation cascade activation [79]. Excessive fibrin within alveoli, coupled with reduced surfactant proteins, can block normal gas exchange, and fibroblast adherence and collagen deposition can promote lung fibrosis which underlie the development of ARDS [80].
Results from a series of 94 COVID-19 patients in Renmin Hospital, Wuhan, China was recently reported [81]. Infected patients showed lower blood levels of antithrombin coupled with higher D-dimer, fibrin/fibrinogen degradation products (FDP), with the highest FDP and D-dimer levels in the more severely ill patients. These results indicated that the blood coagulation cascade has been activated in the COVID-19 patients. A retrospective analysis of 183 patients in Tongji Hospital, Wuhan China, revealed an overall mortality of 11.5% [82]. Upon admission, non-survivors had significantly higher D-dimer, FDP and longer prothrombin time compared with survivors. In a multicenter study of 191 COVID-19 patients in Wuhan, China, 44 of the 54 non-survivors (81%) had a D-dimer level >1 ug/mL upon admission [83]. Activation of the coagulation cascade, as indicated by elevated D-dimer levels, is strongly associated with non-survival in COVID-19 patients.
While most studies to date have originated from China, reports from the growing pandemic in the United States are now emerging. A series of autopsies from New Orleans may shed light on disease progression in the American population [84]. In that series, four patients who died from COVID-19 were studied. The patient ages ranged from 44 to 76. Obesity, hypertension and type II diabetes were common comorbidities as with the Chinese patients. Surprisingly, all the New Orleans patients autopsied were African American.
All four New Orleans patients presented with cough and fever and were hospitalized with respiratory distress. Radiographs indicated bilateral ground glass opacities consistent with ARDS. Laboratory findings included elevated ferritin, fibrinogen, increased prothrombin time, neutrophil count, lymphopenia. In two patients, D-dimer was significantly elevated. Pulmonary pathological findings described small vessels containing small thrombi, fibrin deposition and neutrophil extracellular traps. Thrombotic angiopathy was limited to the lungs.
A similar profile of an extreme immune response coupled with an activated coagulation cascade is associated with non-survival in both the New Orleans and Chinese patients. However, an emerging trend in the United states is the disproportionate number of non-survivors among African American COVID-19 patients. Hemostatic factors previously have been investigated to understand racial/ethnic differences in cardiovascular disease incidence [85]. Black men and women have higher levels of D-dimer than Caucasians, Hispanics or Chinese, whereas Black men have lower levels of plasminogen activator-inhibitor-1 (PAI-1, inhibits fibrinolysis) than other ethnic groups. The genetic basis for these associations is as yet unknown. The high D-dimer levels may indicate a procoagulant state in which the patient is primed for severe COVID-19 disease [86]. Other factors are independently linked to a procoagulant state including obesity [87] and cancer, especially with tumors that express TF [88].
If the formation of microscopic blood clots in the pulmonary microvasculature underlies the development of SARS-CoV-2-induced respiratory failure, and a preexisting procoagulant state promotes this process, anticoagulant therapy might play a role in the treatment of severely ill COVID-19 patients. Upon the basis of abnormal coagulation parameters in severely ill COVID-19 patients, some investigators have suggested that severely ill COVID-9 patients should receive low-molecular weight heparin (LMWH) anticoagulant therapy [89,90]. In a recent clinical study from Tongji Hospital, Wuhan, China, 99 of 449 patients with severe COVID-9 were treated with heparin for at least 7 days [91]. The 28-day mortality for the treated patients was indistinguishable from the untreated patients. However, for patients with high D-dimer levels (> 3 µg/mL), heparin use was associated with increased survival. Immune response and the coagulation cascade have important antiviral activities, however, overactivation of these pathways can also increase disease severity. Future studies will be required to identify which patents should receive anticoagulant therapy and when. The measurement of D-dimer levels should be routine in COVID-19 patients with comorbidities. Collection of this data will be essential for identifying the type and timing of anticoagulant therapy.
The impact of the COVID-19 pandemic is unprecedented. Currently, no FDA approved drugs exist to attenuate the infection in patients, and no vaccine is currently available. Although the SARS-CoV-2 virus was identified only months ago, its similarity to other human pathogenic viruses was immediately apparent. The SARS-CoV-2 RNA sequence revealed several critical viral proteins that are targets for therapy. Substantial prior literature with structurally similar targets has revealed candidate drugs, many of which have been tested already in in vitro models with SARS-CoV-2. Many potential drugs have been used in humans to treat other diseases, and the efficacy of these drugs to treat COVID-19 can be rapidly evaluated.
The clinical presentation of COVID-19 is complex and depends upon age and pre-existing illness. Some but not all patients will progress to life-threatening pneumonia. Identifying those most at risk of progression is necessary. The timing of drug therapy is also a critical and unresolved factor. Sorting out the benefits and toxicities of individual drugs and drug combinations from the numerous and growing clinical trials will be a challenge. As the number of surviving patients grows, it will be critical to understand some of the potentially permanent damage caused by the infection and to incorporate treatment strategies to minimize long term effects. Investments in efforts to answer these questions is essential and will make us more prepared for the next pandemic.
We acknowledge Mariano Garcia-Blanco M.D. Ph.D., Michael Laposata M.D. Ph.D., Rex McCallum M.D. and Wayne Snodgrass M.D. Ph.D. for insightful comments on the manuscript. We thank Mark Sowers, Chia Hsu and James Sowers for valuable comments and assistance with the preparation of the manuscript. This work was supported in part by funds for the John Sealy Distinguished Chair in Cancer Biology and Institute for Translational Sciences at the University of Texas Medical Branch, supported in part by a Clinical and Translational Science Award (UL1 TR001439) from the National Center for Advancing Translational Sciences, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (R24AI120942).
The authors declare no conflicts of interest.
- Chen N, Zhou M, Dong X (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395: 507-513.
- Wang D, Hu B, Hu C (2020) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA - J Am Med Assoc 323: 1061-1069. [Crossref]
- Channappanavar R, Perlman S (2017) Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol 39: 529-539. [Crossref]
- Fu L, Wang B, Yuan T (2020) Clinical characteristics of coronavirus disease 2019 (COVID-19) in China: a systematic review and meta-analysis. J Infect 80: 656-665. [Crossref]
- Liu Y, Du X, Chen J (2020) Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J Infect.
- Liu Z, Long W, Tu M (2020) Lymphocyte subset (CD4+, CD8+) counts reflect the severity of infection and predict the clinical outcomes in patients with COVID-19. J Infect.
- Tan C, Huang Y, Shi F (2020) C-reactive protein correlates with CT findings and predicts severe COVID-19 early. J Med Virol 1-19. [Crossref]
- Ji D, Zhang D, Xu J (2020) Prediction for progression risk in patients with COVID-19 pneumonia: the CALL Score. Clin Infect Dis 1-11.
- Banerjee A, Kulcsar K, Misra V (2019) Bats and coronaviruses. Viruses 11: 7-9.
- Dong S, Sun J, Mao Z (2020) A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019‐nCoV). J Med Virol 0-2.
- Weiss SR, Navas-Martin S (2005) Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 69: 635-664.
- Song Z, Xu Y, Bao L (2019) From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses.
- Hoffmann M, Kleine-Weber H, Schroeder S (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181: 1-10. [Crossref]
- Lukassen S, Lorenz Chua R, Trefzer T (2020) SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J 1-15. [Crossref]
- Harcourt J, Tamin A, Lu X (2020) Severe acute respiratory syndrome coronavirus 2 from patient with 2019 novel coronavirus disease, United States. Emerg Infect Dis 26: 1-8. [Crossref]
- Shang J, Ye G, Shi K (2020) Structural basis of receptor recognition by SARS-CoV-2. Nature 1-8.
- Xie X, Muruato A, Lokugamage KG (2020) An infectious cDNA clone of SARS-CoV-2. Cell Host Microbe 27: 1-8.
- Yamamoto M, Matsuyama S, Li X (2016) Respiratory syndrome coronavirus S protein-mediated membrane. Antimicrob Agents Chemother 60: 6532-6539.
- Wang M, Cao R, Zhang L (2020) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30: 269-271.
- Yamaya M, Shimotai Y, Hatachi Y (2015) The serine protease inhibitor camostat inhibits influenza virus replication and cytokine production in primary cultures of human tracheal epithelial cells. Pulm Pharmacol Ther 33: 66-74. [Crossref]
- Yao X, Ye F, Zhang M (2020) In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome main point : Hydroxychloroquine was found to be more potent than chloroquine at inhibiting SARS-CoV-2 in vit. Clin Infect Dis 2: 1-25.
- Gautret P, Lagier J-C, Parola P (2020) Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents 1-24.
- Schrezenmeier E, Dörner T (2020) Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol 16: 155-166. [Crossref]
- Liu L qing, Wang S bing, Shao Y fei (2019) Hydroxychloroquine potentiates the anti-cancer effect of bevacizumab on glioblastoma via the inhibition of autophagy. Biomed Pharmacother. [Crossref]
- Mackenzie AH (1983) Dose refinements in long-term therapy of rheumatoid arthritis with antimalarials. Am J Med 75: 40-45.
- Marquardt K, Albertson TE (2001) Treatment of hydroxychloroquine overdose. Am J Emerg Med 19: 420-424.
- Rossignol J (2014) Nitazoxanide: A first-in-class broad-spectrum antiviral agent. Antiviral Res 110: 94-103.
- Koszalka P, Tilmanis D, Hurt AC (2017) Influenza antivirals currently in late-phase clinical trial. Influenza Other Respi Viruses 11: 240-246. [Crossref]
- Elazar M, Liu M, McKenna SA (2009) The Anti-Hepatitis C agent nitazoxanide induces phosphorylation of eukaryotic initiation factor 2α via protein kinase activated by double-stranded RNA activation. Gastroenterology 137: 1827-1835.
- Darling JM, Fried MW (2009) Nitazoxanide: Beyond parasites toward a novel agent for hepatitis C. Gastroenterology 136: 760-763.
- Rossignol JF (2016) Nitazoxanide, a new drug candidate for the treatment of Middle East respiratory syndrome coronavirus. J Infect Public Health 9: 227-230.
- Cao J, Forrest JC, Zhang X (2015) A screen of the NIH clinical collection small molecule library identifies potential anti-coronavirus drugs. Antiviral Res 114: 1-10.
- Cao B, Wang Y, Wen D (2020) A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N Engl J Med 1-13.
- Li G, De Clercq E (2020) Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov 19: 149-150.
- Elion GB, Furman PA, Fyfe JA (1977) Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc Natl Acad Sci U S A 74: 5716-5720.
- De Clercq E, Neyts J (2009) Antiviral agents acting as DNA or RNA chain terminators. In: Antivir. Strateg. pp 53-84.
- Huchting J, Vanderlinden E, Van Berwaer R (2019) Cell line-dependent activation and antiviral activity of T-1105, the non-fluorinated analogue of T-705 (favipiravir). Antiviral Res 167: 1-5. [Crossref]
- Jin Z, Smith LK, Rajwanshi VK (2013) The ambiguous base-pairing and high substrate efficiency of T-705 (Favipiravir) Ribofuranosyl 5′-Triphosphate towards influenza A virus polymerase. PLoS One 8: 1-10.
- Huchting J, Winkler M, Nasser H, Meier C (2017) Synthesis of T-705-Ribonucleoside and T-705-Ribonucleotide and studies of chemical stability. ChemMedChem 12: 652-659.
- Vanderlinden E, Vrancken B, Houdt J Van (2016) Distinct effects of T-705 (Favipiravir) and ribavirin on influenza virus replication and viral RNA synthesis. Antimicrob Agents Chemother 60: 6679-6691. [Crossref]
- Siegel D, Hui HC, Doerffler E (2017) Discovery and synthesis of a phosphoramidate prodrug of a Pyrrolo[2,1-f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the treatment of Ebola and emerging viruses. J Med Chem 60: 1648-1661.
- Holshue ML, DeBolt C, Lindquist S (2020) First case of 2019 novel coronavirus in the United States. N Engl J Med 382: 929-936.
- Grein J, Ohmagari N, Shin D (2020) Compassionate use of Remdesivir for patients with severe Covid-19. N Engl J Med 1-10. [Crossref]
- Bassetti M, Castaldo N, Carnelutti A (2019) Neuraminidase inhibitors as a strategy for influenza treatment: pros, cons and future perspectives. Expert Opin Pharmacother 20: 1711-1718.
- Ishaqui AA, Khan AH, Sulaiman SAS (2020) Assessment of efficacy of Oseltamivir-Azithromycin combination therapy in prevention of Influenza-A (H1N1) pdm09 infection complications and rapidity of symptoms relief. Expert Rev Respir Med 00: 1-9.
- Patel VB, Zhong JC, Grant MB, Oudit GY (2016) Role of the ACE2/angiotensin 1-7 axis of the renin-angiotensin system in heart failure. Circ Res 118: 1313-1326.
- Donoghue M, Hsieh F, Baronas E (2000) A novel angiotensin-converting enzyme - related to angiotensin. Circ Res 87: e1-e9.
- Tipnis SR, Hooper NM, Hyde R (2000) A human homolog of angiotensin-converting enzyme: Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275: 33238-33243.
- Li W, Moore MJ, Vasilieva N (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426: 450-454. [Crossref]
- Diaz JH (2020) Hypothesis: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID-19. J Travel Med 1-7. [Crossref]
- Ferrario CM, Jessup J, Chappell MC (2005) Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 111: 2605-2610.
- Schouten LR, van Kaam AH, Kohse F (2019) Age-dependent differences in pulmonary host responses in ARDS: a prospective observational cohort study. Ann Intensive Care.
- Gong Y, Wang Z, Beitelshees AL (2016) Pharmacogenomic Genome-Wide Meta-Analysis of Blood Pressure Response to β-Blockers in Hypertensive African Americans. Hypertension 67: 556-563. [Crossref]
- Chandramohan G, Bai Y, Norris K (2007) Effects of dietary salt on intrarenal angiotensin system, NAD(P)H oxidase, COX-2, MCP-1 and PAI-1 expressions and NF-κB activity in salt-sensitive and -resistant rat kidneys. Am J Nephrol 28: 158-167.
- Williams SF (2014) African Americans, hypertension and the renin angiotensin system. World J Cardiol 6: 878. [Crossref]
- Kobori H, Nishiyama A, Abe Y, Navar LG (2003) Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension 41: 592-597.
- Remková A, Remko M (2010) The role of renin-angiotensin system in prothrombotic state in essential hypertension. Physiol Res 59: 13-23. [Crossref]
- Buckner TW, Key NS (2012) Venous thrombosis in blacks. Circulation 125: 837-839.
- Meng J, Xiao G, Zhang J (2020) Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg Microbes Infect 9: 757-760.
- Vaduganathan M, Vardeny O, Michel T (2020) Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N Engl J Med 1-7.
- Group AIMIC (1998) Indications for ACE Inhibitors in the Early Treatment of Acute Myocardial Infarction. Circulation 97: 2202-2212.
- Pflugfelder PW, Baird G, Tonkon MJ (1993) Clinical consequences of angiotensin-converting enzyme inhibitor withdrawl in chronic heart failure: A double-blind, placebo-controlled study of quinapril. J Am Coll Cardiol 22: 1557-1563. [Crossref]
- Halliday BP, Wassall R, Lota AS (2019) Withdrawal of pharmacological treatment for heart failure in patients with recovered dilated cardiomyopathy (TRED-HF): an open-label, pilot, randomised trial. Lancet 393: 61-73.
- Qiao W, Wang C, Chen B (2015) Ibuprofen Attenuates Cardiac Fibrosis in Streptozotocin-Induced Diabetic Rats. Cardiology 131: 97-106.
- Day M (2020) Covid-19: ibuprofen should not be used for managing symptoms, say doctors and scientists. BMJ 368: m1086.
- Walsh P, Lebedev M, McEligot H (2020) A randomized controlled trial of a combination of antiviral and nonsteroidal anti-inflammatory treatment in a bovine model of respiratory syncytial virus infection. PLoS One 15: 1-17.
- Fitzgerald GA (2020) Misguided drug advice for COVID-19. Science 367: 1434-1436.
- Amici C, Di Caro A, Ciucci A (2006) Indomethacin has a potent antiviral activity against SARS coronavirus. Antivir Ther 11: 1021-1030.
- Shacter E, Lopez RL, Pati S (1991) Inhibition of the myeloperoxidase-H2O2-Cl- system of neutrophils by indomethacin and other non-steroidal anti-inflammatory drugs. Biochem Pharmacol 41: 975-984.
- Culic O, Erakovic V, Cepelak I (2002) Azithromycin modulates neutrophil function and circulating inflammatory mediators in healthy human subjects. Eur J Pharmacol 450: 277-289. [Crossref]
- Tamaoki J, Tagaya E, Sakai A, Konno K (1995) Effects of macrolide antibiotics on neurally mediated contraction of human isolated bronchus. J Allergy Clin Immunol 95: 853-859.
- Shinkai M, Henke MO, Rubin BK (2008) Macrolide antibiotics as immunomodulatory medications: Proposed mechanisms of action. Pharmacol Ther 117: 393-405.
- Michot J-M, Albiges L, Chaput N (2020) Tocilizumab, an anti-IL6 receptor antibody, to treat Covid-19-related respiratory failure: a case report. Ann Oncol.
- Luo P, Liu Y, Qiu L (2020) Tocilizumab treatment in COVID-19: a single center experience. J Med Virol 1-10. [Crossref]
- Pawar A, Desai RJ, Solomon DH (2019) Risk of serious infections in tocilizumab versus other biologic drugs in patients with rheumatoid arthritis: A multidatabase cohort study. Ann Rheum Dis 78: 456-464.
- Antoniak S, Mackman N (2014) Coagulation, Protease Activated Receptors and Viral Myocarditis. J Cardiovasc Transl Res 7: 203-211.
- Antoniak S (2018) The coagulation system in host defense. Res Pract Thromb Haemost 2: 549-557.
- Leavell KJ, Peterson MW, Gross TJ (1996) The Role of Fibrin Degradation Products in Neutrophil Recruitment to the Lung. Am J Respir Cell Mol Biol 14: 53-60. [Crossref]
- Gralinski LE, Baric RS (2015) Molecular pathology of emerging coronavirus infections. J Pathol 235: 185-195. [Crossref]
- Gralinski LE, Iii AB, Jeng S (2013) Mechanisms of Severe Acute Respiratory Syndrome Coronavirus-Induced Acute Lung Injury. MBio 4: 1-12.
- Han H, Yang L, Liu R (2020) Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med.
- Tang N, Li D, Wang X, Sun Z (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 18: 844-847. [Crossref]
- Zhou F, Yu T, Du R (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395: 1054-1062.
- Fox SE, Akmatbekov A, Harbert JL (2020) Pulmonary and Cardiac Pathology in Covid-19: The First Autopsy Series from New Orleans. medRxiv 1-8.
- Lutsey PL, Cushman M, Steffen LM (2006) Plasma hemostatic factors and endothelial markers in four racial/ethnic groups: The MESA study. J Thromb Haemost 4: 2629-2635.
- Khaleghi M, Saleem U, McBane RD (2009) African-American ethnicity is associated with higher plasma levels of D-dimer in adults with hypertension. J Thromb Haemost 7: 34-40. [Crossref]
- Eichinger S, Hron G, Bialonczyk C (2008) Overweight, obesity, and the risk of recurrent venous thromboembolism. Arch Intern Med 168: 1678-1683. [Crossref]
- Streiff MB (2013) Association between cancer types, cancer treatments, and venous thromboembolism in medical oncology patients. Clin Adv Hematol Oncol 11: 349-357. [Crossref]
- Li T, Lu H, Zhang W (2020) Clinical observation and management of COVID-19 patients. Emerg Microbes Infect 9: 687-690.
- Lin L, Lu L, Cao W, Li T (2020) Hypothesis for potential pathogenesis of SARS-CoV-2 infection-a review of immune changes in patients with viral pneumonia. Emerg Microbes Infect 9: 727-732.
- Tang N, Bai H, Chen X (2020) Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 1-15. [Crossref]