Abstract
Alzheimer's disease (AD) is a debilitating neuropsychiatric disorder characterized by the multifaceted decline in cognitive and behavioral functions. Due to the multifaceted nature of AD pathology and our limited understanding on its etiology, AD is difficult to be treated with currently available pharmaceuticals. Then, new therapeutic strategies for AD have been proposed. Since 1975, several clinical studies have reported that L-type Ca2+ channel blockers (CCBs) used in anti-hypertensive therapy produces increase of plasma catecholamine levels and tachycardia, typical symptoms of sympathetic hyperactivity. Despite these adverse effects of CCBs have been initially attributed to adjust reflex of arterial pressure, during almost four decades these enigmatic phenomena remained unclear. In 2013, we discovered that this paradoxical sympathetic hyperactivity produced by CCBs results from the increase of catecholamines release from sympathetic nerves and adrenal chromaffin cells due to its modulatory action on the interaction between intracellular signaling pathways mediated by Ca2+ and cAMP (Ca2+/cAMP signalling interaction). In addition, we discovered that the modulation of this interaction may stimulate neuroprotective response due to activation of cell survival pathways mediated by cAMP-responsive element binding protein (CREB). Then, the pharmacological modulation of Ca2+/cAMP signalling interaction by combined use of L-type CCBs and cAMP-enhancer compounds could be a more efficient and safer therapeutic strategy to produce increase of cholinergic neurotransmission and neuroprotection, attenuating cognitive deficit in AD patients.
Key words
Ca2+/cAMP signalling interaction, neurotransmission, neuroprotection, Alzheimer´s disease.
Introduction
Since 1970´s, several clinical studies have reported that acute and chronic administration of L-type Ca2+ channel blockers (CCBs) in hypertensive patients, such as nifedipine and verapamil, decreased arterial pressure but produced typical symptoms of sympathetic hyperactivity such as tachycardia, and increment of catecholamine plasma levels [1]. Despite these adverse effects of CCBs have been initially credited to adjust reflex of arterial pressure, the cellular and molecular mechanisms involved in these CCBs-effects remained unclear for decades. Our previous studies performed in isolated tissues richly innervated by sympathetic nerves (rat vas deferens), to exclude the influence of adjusting reflex, showed that neurogenic responses were completely inhibited by L-type CCBs in high concentrations (>1 μmol/L), but unexpectedly and paradoxically potentiated in concentrations below 1 μmol/L, characterizing CCBs-induced sympathetic hyperactivity [2-4]. During almost four decades, these paradoxical effects of CCBs named by us as “calcium paradox” remained unclear.
In 2013, we discovered that this paradoxical sympathetic hyperactivity produced by L-type CCBs is due to its modulatory action on the interaction between the intracellular signalling pathways mediated by Ca2+ and cAMP (Ca2+/cAMP signalling interaction). Our studies have showed that the pharmacological modulation of the Ca2+/cAMP signalling interaction by combined use of L-type CCBs and compounds which increase cytosolic cAMP concentration ([cAMP]c), named cAMP-enhancer compounds, could be useful to increase neurotransmission, and neuroprotection in neurological and psychiatric disorders, such as Alzheimer´s diseases (AD) [5-8].
Current therapy to treat Alzheimer´s disease (AD)
Accumulation of the β-amyloid peptide (Aβ) in brain tissues represents the pathological status of AD [9,10]. According to the amyloid hypothesis, disruption of homeostatic processes causes overproduction of Aβ. Age-related factors could favor a metabolic alteration, favoring the amyloidogenic processing [9,10]. The neurotoxic potential of the Aβ results from its potential to favor aggregation. This process, along with a reduction of Aβ removal from the brain, leads to the extracellular accumulation of Aβ, and the subsequent activation of neurotoxic cascades, that ultimately leads to neuronal dysfunction and cellular death [9,10]. The relevance of the early diagnosis of AD relies on the hypothesis that pharmacological interferences on disease-modifying complexes are more likely to produce clinically relevant rules if started early enough in the continuum towards dementia [9,10]. Therapies leading the change of amyloid-related cascades may be viewed as promising plans to attenuate or even to revert dementia [10]. Therefore, the cumulative knowledge on the pathogenesis of AD derived from basic science models will hopefully be translated into clinical practice in the upcoming years. Other targets relevant to AD have also been considered in the last years for making multitarget compounds [11,12].
In addition to what has been discussed above, acetylcholinesterase (AChE) is another important target to treat the pathogenesis of AD (cholinergic dysfunction hypothesis). Considering the current hypothesis of accretion of the Aβ in AD, this relies in the reduction of acetylcholine release in central cholinergic nervous system involved in cognitive function. Thus, the inhibition of acetylcholine degradation by AChE is a potential goal to treat AD [11,12]. Deleterious excess Ca2+ influx is also another constituent seen in aging and neurodegenerative diseases [13]. Thus, hybrid compounds having the moieties of tacrine (potent inhibitor of brain and peripheral AChE), and nimodipine (L-type CCBs) have been synthetized [11,12]. In addition, galantamine, a mild AChE inhibitor, and an allosteric ligand of nicotinic receptors, has been used to progress enhancing cognition and behavior in patients with AD [14]. Finally, N-methyl-D-aspartat (NMDA) receptor antagonist (memantine) has also been proposed to treat this disease [15]. Besides the current medicines available nowadays in clinics, new insights for more efficient pharmacological treatments of AD are clearly needed.
Role of the Ca2+/cAMP signalling interaction in neurotransmission
Many experiments studies initiated decades ago, using adrenal chromaffin cells as cellular model, established the notion of stimulus-secretion coupling to explain transmitter release from central and peripheral neurons. In 1970´s, it was discovered that a rise in the cytosolic Ca2+ concentration ([Ca2+]c) is an elementary requirement to trigger transmitter release from adrenal chromaffin cells [16]. In 1990´s, it was showed a direct relationship between rise in [Ca2+]c and rapid transmitter release from adrenal chromaffin cells [17]. It was also showed that increase of [cAMP]c in adrenal chromaffin cells due to activation of adenylate cyclase by forskolin enhances release of secretory vesicles containing transmitters (catecholamines, purines and other substances) [18]. These findings support that both Ca2+ and cAMP are involved in the regulation of neurotransmitter release at many peripheral and central synapses of mammals, including sympathetic synapses.
In 2013, we discovered that neurotransmitter release from sympathetic neurons is finely regulated by interaction between intracellular signalling pathways mediated by Ca2+ and cAMP, named Ca2+/cAMP signalling interaction [6]. In fact, the hypothesis for a suitable Ca2+/cAMP signalling interaction has been widely studied in different cell types and tissues. This interaction results in synergistic actions of these intracellular messengers on cell functions regulated by adenylyl cyclases (ACs), or phosphodiesterases (PDEs) [5-8]. The Ca2+/cAMP signalling interaction has particularly been extensively studied at the endoplasmic reticulum (ER) Ca2+ channels, such as ER-Ca2+ channels regulated by ryanodine receptors (RyR) [5-8]. Our studies established that Ca2+/cAMP signalling interaction play an important role in neurotransmitter release regulation in neurons and neuroendocrine cells [5-8]. Thus, pharmacological modulation of this interaction produced by L-type CCBs and cAMP-enhancer drugs could be useful to treat neurological and psychiatric disorders resulting from neurotransmitter release deficit, such as AD, Parkinson's disease and depression [5-8].
Role of the Ca2+/cAMP signalling interaction in neuroprotection
It is well established that cytosolic Ca2+ overload is directly involved in neuronal death in various neurodegenerative diseases, including AD and Parkinson's diseases [5-8]. Recently, it was showed that the treatment with L-type CCBs, such as isradipine, reduces motor symptoms and attenuates progressive death of dopamine neurons from substantia nigra in animal model of Parkinson's disease [19]. It was showed that isradipine produces a dose-dependent sparing of dopaminergic fibers, and cell bodies at concentrations achievable in humans [20], suggesting that L-type CCBs are potentially viable neuroprotective agents for Parkinson's disease. A phase II clinical trial published in 2016 showed that treatment with isradipine was safely tolerated to reduce motor symptoms by patients with Parkinson's disease [20]. In addition, a 10-year follow-up study (2000 to 2010), involving 82,107 hypertensive patients of more than 60 years of age, showed that use of L-type CCBs reduced blood pressure, and risk of dementia in hypertensives patients, suggesting that these drugs could be clinically used to treat AD [21]. These findings reinforced the idea that attenuation of cytosolic Ca2+ overload produced by L-type CCBs due to blockade of Ca2+ influx through L-type voltage-activated Ca2+ channels (VACC) could be an excellent pharmacological strategy to attenuate, or prevent, neuronal death in neurodegenerative diseases, such as AD and Parkinson's disease.
As previously mentioned, blockade of the L-type VACC by CCBs reduces Ca2+ influx and [Ca2+]c, increasing ACs activity and [cAMP]c [5-8]. This functional Ca2+/cAMP signalling interaction regulates various cellular responses, including neurotransmitter release [5-8]. Many studies showed that increase of [cAMP]c stimulates neuroprotective response attenuating neuronal death due probably to activation of cellular survival pathways mediated by cAMP-response element binding protein (CREB) [22-24]. In this way, pharmacological modulation of the Ca2+/cAMP signalling interaction by combined use of L-type CCBs and cAMP-enhancer compounds could stimulate neuroprotective response due to increase of [cAMP]c and attenuation of cytosolic Ca2+ overload [5-8]. Thus, pharmacological modulation of this interaction could be a new neuroprotective therapeutic strategy to slow the progression of neurodegenerative diseases, such as AD and Parkinson's disease.
Pharmacological modulation of neural Ca2+/cAMP signalling interaction as therapeutic goal for treatment of Alzheimer´s disease (AD)
Our discovery of the involvement of the Ca2+/cAMP signalling interaction in the neurotransmission, and neuroprotection, has produced important advances in the understanding of the pathophysiology and pharmacology of neurological and psychiatric disorders [5-8]. These advances allowed us to propose that pharmacological modulation of the Ca2+/cAMP signalling interaction produced by combined use of the L-type CCBs such as isradipine (usually indicated in antihypertensive therapy), and cAMP-enhancer compounds such as rolipram (usually indicated in anti-depressive therapy), could represent a new therapeutic strategy for enhancing neurotransmission, and producing neuroprotection in the neurodegenerative diseases, such as AD.
Our studies suggest that pharmacological modulation of the Ca2+/cAMP signalling interaction by combined use of the L-type CCBs and cAMP-enhancer compounds induces enhance of neurotransmission due to increase of neurotransmitter release mediated by Ca2+ release from ER stimulated by cAMP [5-8]. This Ca2+ release from ER produces increase number of secretory vesicles docked in plasma membrane, increasing neurotransmitter release [5-8]. Then, pharmacological modulation of the Ca2+/cAMP signalling interaction could be a new therapeutic strategy to treat neurological and psychiatric disorders resulting from neurotransmitter release deficit. In addition, pharmacological modulation of the Ca2+/cAMP signalling interaction could reduce neuronal death in neurodegenerative disease due to attenuation of cytosolic Ca2+ overload, increase of [cAMP]c and stimulation of cell survival pathways mediated by CREB [22-24]. Thus, pharmacological modulation of Ca2+/cAMP signalling interaction could be a new neuroprotective therapeutic strategy to slow the progression of neurodegenerative diseases, such as AD [5-8,25,26].
Our proposal of pharmacological modulation of the Ca2+/cAMP signalling interaction could open a new avenue for the drug development more effective and safer to reduce clinical symptoms of neurological and psychiatric disorders resulting from neurotransmitter release deficit, and neuronal death triggered by cytosolic Ca2+ overload, such as AD [5-8,25,26]. Figure 1 show how pharmacological modulation of the Ca2+/cAMP signalling interaction using L-type CCBs and cAMP-enhancer compounds could produce increase of neurotransmission and neuroprotection.
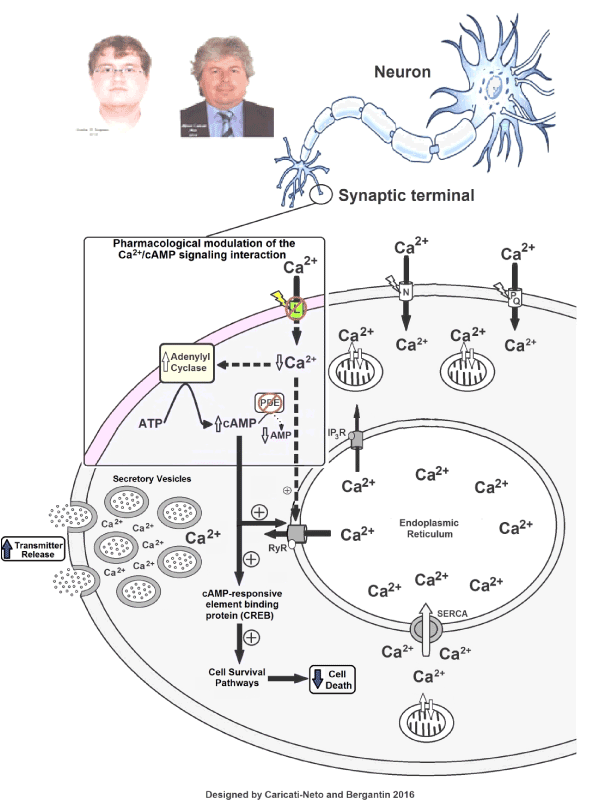
Figure 1. Increase of neurotransmitter release and attenuation of neuronal death produced by pharmacological modulation of the Ca2+/cAMP signalling interaction by combined use of L-type Ca2+ channel blockers (CCBs) and cAMP-enhancer compounds, such as phosphodiesterase (PDE) inhibitors.
Conclusion
Our recent discovery of role of the Ca2+/cAMP signalling interaction in the neurotransmission and neuroprotection could promote important advances in the pathophysiology and pharmacology of the neurological and psychiatric disorders. These advances can contribute to drug development more effective and safer to attenuate clinical symptoms of neurological and psychiatric disorders, such as AD.
Disclosure statement
Caricati-Neto and Bergantin thank the continued financial support from CAPES, CNPq and FAPESP (Bergantin´s Postdoctoral Fellowship FAPESP #2014/10274-3).
References
- Grossman E, Messerli FH (1998) Effect of calcium antagonists on sympathetic activity. Eur Heart J 19 Suppl F: F27-31. [Crossref]
- Kreye VA, Luth JB (1975) Proceedings: verapamil-induced phasic contractions of the isolated rat vas deferens. Naunyn Schmiedebergs Archives of Pharmacology 287 (Suppl R): R43. [Crossref]
- French AM, Scott NC (1981) A comparison of the effects of nifedipine and verapamil on rat vas deferens. Br J Pharmacol 73: 321-323. [Crossref]
- Moritoki H, Iwamoto T, Kanaya J, Maeshiba Y, Ishida Y, et al. (1987) Verapamil enhances the non-adrenergic twitch response of rat vas deferens. European Journal of Pharmacology 140: 75–83. [Crossref]
- Caricati-Neto A, García AG, Bergantin LB (2015) Pharmacological implications of the Ca2+/cAMP signalling interaction: from risk for antihypertensive therapy to potential beneficial for neurological and psychiatric disorders. Pharmacology Research and Perspectives 3: e00181. [Crossref]
- Bergantin LB, Souza CF, Ferreira RM, Smaili SS, Jurkiewicz NH, et al. (2013) Novel model for "calcium paradox" in sympathetic transmission of smooth muscles: role of cyclic AMP pathway. Cell Calcium 54: 202-212. [Crossref]
- Bergantin LB, Jurkiewicz A, García AG, Caricati-Neto A (2015) A Calcium Paradox in the Context of Neurotransmission. Journal of Pharmacy and Pharmacology 3: 253-261. [Crossref]
- Bergantin LB, Caricati-Neto A (2016) Challenges for the pharmacological treatment of neurological and psychiatric disorders: Implications of the Ca2+/cAMP intracellular signalling interaction. European Journal Pharmacology 788: 255-260. [Crossref]
- De-Paula VJ, Radanovic M, Diniz BS, Forlenza OV (2012) Alzheimer's disease. Subcell Biochem 65: 329-352. [Crossref]
- Mohamed T, Shakeri A, Rao PP (2016) Amyloid cascade in Alzheimer's disease: Recent advances in medicinal chemistry. Eur J Med Chem 113: 258-272. [Crossref]
- Ismaili L, Refouvelet B, Benchekroun M, Brogi S, Brindisi M, et al. (2016) Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer's disease. Prog Neurobiol 151: 4-34. [Crossref]
- Maroto M, de Diego 2021 Copyright OAT. All rights reservles JC, Caricati-Neto A, et al. (2011) Multi-target novel neuroprotective compound ITH33/IQM9.21 inhibits calcium entry, calcium signals and exocytosis. Cell Calcium 50: 359–369. [Crossref]
- Kawamoto EM, Vivar C, Camandola S (2012) Physiology and pathology of calcium signaling in the brain. Front Pharmacol 3: 61. [Crossref]
- Samochocki M, Höffle A, Fehrenbacher A, Jostock R, Ludwig J, et al. (2003) Galantamine is an allosterically potentiating ligand of neuronal nicotinic but not of muscarinic acetylcholine receptors. J Pharmacol Exp Ther 305: 1024-1036. [Crossref]
- Devi L, Ohno M (2016) Cognitive benefits of memantine in Alzheimer's 5XFAD model mice decline during advanced disease stages. Pharmacol. Biochem Behav 144: 60-66. [Crossref]
- Baker PF, Knight DE (1978) Calcium-dependent exocytosis in bovine adrenal medullary cells with leaky plasma membranes. Nature 276: 620–622. [Crossref]
- Neher E, Zucker RS (1993) Multiple calcium-dependent processes related to secretion in bovine chromaffin cells. Neuron 10: 21-30. [Crossref]
- Chern YJ, Kim KT, Slakey LL, Westhead EW (1988) Adenosine receptors activate adenylate cyclase and enhance secretion from bovine adrenal chromaffin cells in the presence of forskolin. Journal of Neurochemistry 50: 1484–1493. [Crossref]
- Ilijic E, Guzman JN, Surmeier DJ (2011) The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis 43: 364-371. [Crossref]
- Swart T, Hurley MJ (2016) Calcium channel antagonists as disease-modifying therapy for Parkinson's disease: Therapeutic rationale and current status. CNS Drugs 30:1127-1135. [Crossref]
- Wu CL, Wen SH (2016) A 10-year follow-up study of the association between calcium channel blocker use and the risk of dementia in elderly hypertensive patients. Medicine (Baltimore) 95: e4593. [Crossref]
- Sommer N, Loschmann PA, Northoff GH, Weller M, Steinbrecher A, et. al. (1995) The antidepressant rolipram suppresses cytokine production and prevents autoimmune encephalomyelitis. Nature Medicine 1: 244–248. [Crossref]
- Xiao L, O'Callaghan JP, O'Donnell JM (2011) Effects of repeated treatment with phosphodiesterase-4 inhibitors on cAMP signaling, hippocampal cell proliferation, and behavior in the forced-swim test. Journal of Pharmacology Experimental and Therapeutics 338: 641–647. [Crossref]
- Li Y, Wu KJ, Yu SJ, Tamargo IA, Wang Y, Greig NH (2016) Neurotrophic and neuroprotective effects of oxyntomodulin in neuronal cells and a rat model of stroke. Experimental Neurology 288: 104-113. [Crossref]
- Bergantin LB, Caricati-Neto A (2016) Insight from “Calcium Paradox” due to Ca2+/cAMP Interaction: Novel Pharmacological Strategies for the Treatment of Depression. Int Arch Clin Pharmacol 2: 007. [Crossref]
- Bergantin LB, Caricati-Neto A (2016) Novel Insights for Therapy of Parkinson’s disease: Pharmacological Modulation of the Ca2+/cAMP Signalling Interaction. Austin Neurol & Neurosci 1: 1009. [Crossref]