To insight the pathogenesis of Mesangial proliferative glomerulonephritis (MsPGN), we investigated the phosphoproteomic profile of PBMCs from MsPGN patients and normal subjects by integrating TiO2 enrichment technology, 2D nano-liter liquid chromatography and linear ion trap quadrupole mass spectrometry. We identified totally 693 differential phosphorylation sites and corresponded to 439 genes. Gene ontology (GO) analysis showed that protein or nucleic acid binding took up the largest proportion of molecular function, followed by nucleobase, nucleoside, nucleotide and nucleic acid metabolic process in the nucleus. KEGG Pathway analysis showed that most of differential gene enrich in mitogen-activated protein kinase (MAPK) signaling pathway and focal adhesion pathway. Gene network analysis showed that serine/arginine repetitive matrix (SRRM) 1, histone deacetylase (HDAC) 1 and protein kinase C delta (PRKED) were significantly regulators in the network. These results suggested that abnormal changes of protein phosphorylation modification may contribute to MsPGN, and may be derived from the dysregulation of MAPK signaling pathway and focal adhesion pathway. In these pathways, the differential genes SRRM1, HDAC1 and PRKCD with higher connection may be the promising biomarker for MsPGN.
mesangial proliferative glomerulonephritis, differential phosphorylation sites, molecular function, signal pathway
Mesangial proliferative glomerulonephritis is one of the most frequent forms of glomerulonephritis, characterized by mesangial hypercellularity and glomerular extracellular matrix expansion, often accompanied by mononuclear cell infiltration [1-3]. Generally, a renal biopsy is necessary for an accurate diagnosis to observe the lesions in a kidney of MsPGN patients [4]. However, due to the mechanical invasion with the risk of infection and hemorrhage, it is not very suitable for a damaged renal to repeat this approach [5]. Therefore, the development of new biomarkers would be of great benefit for further understanding the pathogenesis and improving the diagnosis, prognosis and treatment of MePGN. Proteomic has been used to globally analyze intracellular signaling events as well as related post-translational modifications. Few studies have been reported the complicated alteration of proteomics in a variety of diseases, even in MePGN [6-8]. W Sui, et al. [8] utilized iTRAQ technology to analyze the total proteins in renal tissues of MePGN patients, and found that differentially expressed proteome profiles of MePGN may enable the development of new approaches to diagnosis of MePGN patients.
phosphorylation is a complex regulatory event which is involved in the biological activity of protein that affect virtually the signaling networks [9]. It has been confirmed that phosphorylation is associated with Systemic lupus erythematosus Alzheimer's disease Cysticfibrosis and Severe combined immunodeficiency [10-12]. However, an integrated analysis of the phosphoproteome in patients with MsPGN has been rarely reported in the worldwide. Thus, the research of proteomics would be of great benefit to find new biomarkers for further understanding the etiology and improving the diagnosis of MsPGN. The phosphopeptide proteome utilized highly sensitive Liquid chromatography-mass spectrometry (LCMS/MS) system, improved software for phosphopeptide identification and subsequent analysis with an elaborate bioinformatics strategy, including gene ontology (GO) analysis, pathway analysis and protein network analysis [10]. The rich data from the proteomic analysis also provides insight into the pathogenesis of MsPGN.
In this study, to investigate the significance of phosphoproteome in MePGN, we used a method that combines TiO2 enrichment, 2D nano-liter LCMS/MS to explore the differentially expressed phosphoproteome in MsPGN patients, analyzed their gene ontology and pathway at the same time.
PBMCs culture and lysis
This study enrolled 12 subjects which included 6 MsPGN patients and 6 healthy volunteers (Table 1). All MsPGN patients were recruited from the inpatient in nephrology department of Guilin 181st hospital and were free of active infections, diabetes mellitus, and autoimmune diseases. None of these patients received immunosuppressive treatment or nonsteroidal anti-inflammatory drugs during this study. All cases were primary chronic glomerulonephritis. The immune deposition was homogeneous such as deposition of IgA and C3 be associated with faint deposition of IgG. Age-, race-, and sex- matched healthy controls were recruited in physical examination department. The Ethics Committee of Guilin 181st hospital approved the study and peripheral blood samples were obtained with informed consent from all participating individuals. Under clinical aseptic conditions, peripheral blood samples of 6 MsPGN patients and 6 normal were phlebotomized individually. All isolated PBMCs were separated by lymphocyte separation medium (Lympholyte®-H), then gathered in EP tube separately. PBMCs was cultured essentially according to American Type Culture Collection (ATCC) recommendations. PBMCs were cultured for 24 hours in 24-well plates, using supplemented RPMI-1640 medium, and incubated at 37℃in a humidified, 5% CO2 atmosphere. PBMCs proliferate with phytohemagglutinin (PHA) stimulus. All PBMCs were eluted twice in ice-cold buffer solution. Then protein concentrations were measured by bicinchoninic acid (BCA) assay.
Table 1. Baseline clinical data of patients and controls
|
|
MsPGN patients
|
Healthy controls
|
|
Males/females (n)
|
4/2
|
4/2
|
|
Age (years)
|
37.75±17.76
|
35.50±4.93
|
|
Systolic (mmHg)
|
137.25±20.12
|
118.33±7.26
|
|
Diastolic (mmHg)
|
88.00±5.83
|
83.17±8.11
|
|
Hemoglobin (g/L)
|
117.25±34.71
|
98.83±14.43
|
|
Total protein (g/L)
|
42.50±10.86
|
77.99±8.14
|
|
Albumin (g/L)
|
20.60±5.23
|
53.16±12.54
|
|
Globulin (g/L)
|
21.90±9.81
|
12.58± 1.99
|
|
Creatinine (µmol/L)
|
105.75±43.21
|
84.67±19.54
|
|
Uric acid (µmol/L)
|
401.75±67.39
|
236.00± 55.57
|
|
Urea (mmol/L)
|
7.53±6.07
|
17.07±10.89
|
|
Calcium (mmol/L)
|
1.86±0.14
|
2.08± 0.37
|
|
Phosphorus (mmol/L)
|
1.42±0.33
|
1.15±0.44
|
|
Kalium (mmol/L)
|
3.65±0.25
|
2.53±0.49
|
Phosphopeptide enrichment
When cells reached suitable confluence, we dissolved cells via two-step LysC and trypsin digestion in the presence of SDS. Phosphopeptides were enriched by TiO2 bead. This phosphopeptide enrichment strategies for high-resolution mass spectrometry had a renaissance in phosphorylation sites mapping, and recent large-scale studies have strengthened the content of phosphorylation. Briefly, peptide samples were primarily incubated with TiO2 beads, then eluted in NH4OH. Each enriched eluent was gathered for mass spectrometric analysis. Subsequently, the remaining part of the eluent was into several fractions by IEF using the Agilent 3100 OFFGEL Fractionator. We added 10 µl of 10% TFA to each fraction and stage-tipped. Peptides were collected at a maximum current of 50 µA and maximum power of 200 mW [12]. Finally, all eluates in TiO2 column were separated, concentrated and dried and stored at -80°C for future use.
Mass spectrometry: We performed mass spectrometry using a 2D nano-liter liquid chromatography system coupled to a linear ion trap quadrupole mass spectrometry (LTQ-Orbitrap XL, Thermo Fisher Scientific, San Jose, CA). The process of LTQ-Orbitrap XL consisted of MS1 scan and MS2 scans. We collected peptides and measure peptide fragments generated and used automatic gain control target values of 1000000 for MS1 and 50000 for MS2. Dynamic exclusion was activated for this process, with a repeat count, exclusive duration of 20 s and ±5 ppm mass tolerance. MS1 scan was profile mode, MS2 scan was centroid mode and multistage activation.
False discovery rate (FDR): Raw files were preprocessed using MaxQuant 1.0.13.13. In MaxQuant, we estimated false discovery rate (FDR) of all peptide identifications at 1% via automatically filtering according to peptide length, mass error estimates, and Mascot scores of all forward and reversed peptide identifications. The spectra were identified via International Protein Index (IPI; version 3.52) human database and the comprehensive enzyme specificity via Open Mass Spectrometry Search Algorithm (OMSSA; version 2.1.4). The intensity of phosphopeptides were identified using Mascot, and Coon OMSSA Proteomic Analysis Software Suite (COMPASS) was utilized to screen peptide, protein and phosphorylation site to FDR ≤ 0.01.
Statistical analysis: For screening differential phosphorylation sites between two groups, following methods were used. ① Calculated the fold change between two groups. ② Set threshold value as 1, which signified the average fold change between two groups was ≥ 2 folds; and the p value of t-test was ≤ 0.05. T-test was conducted using MATLAB 7.5. ③ Labeled gene names corresponding proteins according to the NCBI database.
Differential phosphorylation sites identification and cluster analysis
In this study, the phosphoproteome in PBMCs of MsPGN was profiled using an ultrasensitive and high resolution mass spectrometer. With less than two weeks, we utilized euclidean distance and average to analyze the cluster of differential phosphorylation sites and identified 693 differential phosphorylation sites and 439 corresponding genes altogether according to the information which was remarked in NCBI database.
GO analysis
To characterize the phosphoproteome, the single or multiple biological functions associated with differential phosphoproteins was identified via GO database. The major functional informations of GO were biological process, cellular component and molecular function. The results indicated that nucleotide and nucleic acid metabolic process (21.4%), cellular component organization (21.3%), transport (14.7%) and multicellular organismal development (12.1%) (Figure 1A) were prominent in biological processes. Cellular component classification showed that nucleus (29.7%), plasma membrane (16.6%), cytosol (15.8%) and cytoskeleton (13.1%) were dominant subcellular components represented in phosphorylation (Figure 1B). The most important molecular functions involved in phosphorylation were protein binding (35.5%), catalytic activity (16.1%), nucleic acid binding (13.6%) and nucleotide binding (12.9%) in PBMCs of MsPGN (Figure 1C).
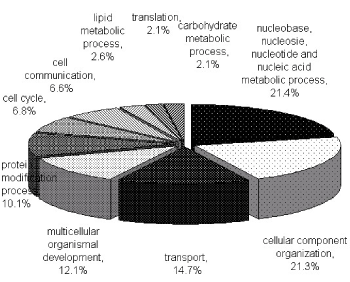
Figure 1A. Functional classification of phosphoproteins in PBMCs based on the involvement in biological processes. The largest group contains proteins related to nucleobase, nucleoside, nucleotide and nucleic acid metabolism process. Two other large groups are the proteins involved in cellular component organization and transport
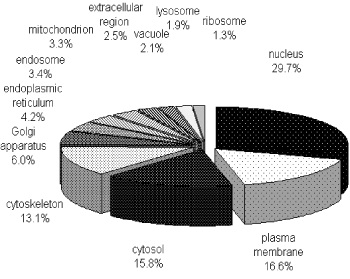
Figure 1B. Distribution of phosphoproteins based on the location of cellular components. The most enriched cellular components were nuclear proteins and proteins associated with the plasma membrane, cytosol or cytoskeleton. The information was compiled from Gene Ontology annotations
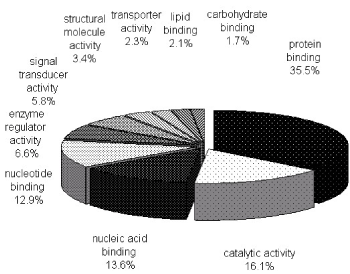
Figure 1C. Molecular functions of phosphoproteins in PBMCs. The largest group is constituted by protein binding followed by catalytic activity and nucleic acid binding. The information was compiled from Gene Ontology annotations
Metabolism comprised a variety of signaling pathways, which are activated by DNA, RNA alteration and replication stress, and transduced by kinase cascades, mainly through some specific protein kinases. Canonical pathway mapping was performed using GenMAPP v2.1 through KEGG pathway database. This study identified 44 metabolic pathways associated with most differential phosphorylation sites (Table 2), which contained MAPK signaling pathway, focal adhesion, regulation of actin cytoskeleton, FC gamma R-mediated phagocytosis, vascular smooth muscle contraction, RNA transport and so on. MAPK signaling pathway (Figure 2A) and focal adhesion (Figure 2B) appears higher rate which demonstrated more alteration in these pathways.
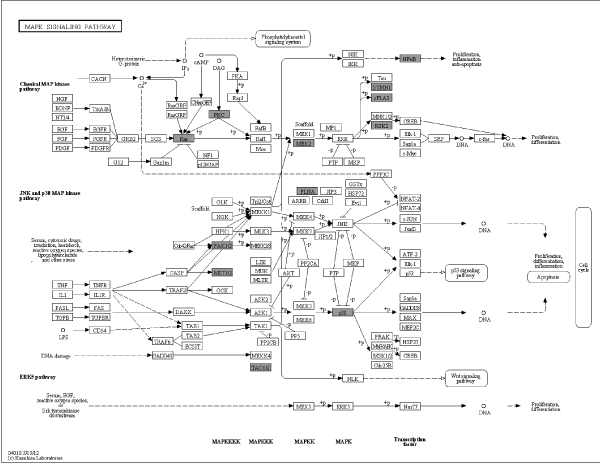
Figure 2A. Pathway analysis in PBMCs of MePGN. Specific content of MAPK signaling pathway was shown, black shadow rectangles symbolized differentially expressed genes
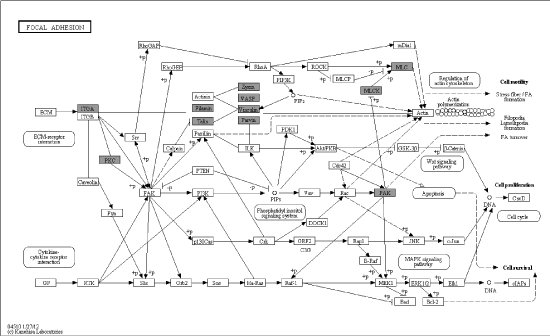
Figure 2B. Details of focal adhesion based on pathway analysis, black shadow rectangles symbolized differentially expressed genes
Table 2. Rank of differentially-expressed pathways in MsPGN patients versus healty controls
|
Pathway
|
P value
|
|
MAPK signaling pathway
|
0.003494
|
|
Focal adhesion
|
0.000573
|
|
Fc gamma R-mediated phagocytosis
|
8.16E-07
|
|
Vascular smooth muscle contraction
|
7.14E-06
|
|
Regulation of actin cytoskeleton
|
0.002977
|
|
RNA transport
|
0.000656
|
|
Fc epsilon RI signaling pathway
|
5.77E-06
|
|
GnRH signaling pathway
|
4.86E-05
|
|
T cell receptor signaling pathway
|
9.03E-05
|
|
Leukocyte transendothelial migration
|
0.000172
|
|
Tight junction
|
0.000534
|
|
Chemokine signaling pathway
|
0.00902
|
|
Endocytosis
|
0.034331
|
|
Pancreatic secretion
|
0.001066
|
|
Natural killer cell mediated cytotoxicity
|
0.009163
|
|
Herpes simplex infection
|
0.043805
|
|
Long-term depression
|
0.000385
|
|
Gastric acid secretion
|
0.000563
|
|
Phosphatidylinositol signaling system
|
0.000951
|
|
Salmonella infection
|
0.001529
|
|
Salivary secretion
|
0.001907
|
|
Ribosome
|
0.002198
|
|
Spliceosome
|
0.015999
|
|
Long-term potentiation
|
0.001978
|
|
Progesterone-mediated oocyte maturation
|
0.006306
|
|
Gap junction
|
0.007588
|
|
Retrograde endocannabinoid signaling
|
0.013264
|
|
Glutamatergic synapse
|
0.042641
|
|
Shigellosis
|
0.004493
|
|
Epithelial cell signaling in Helicobacter pylori infection
|
0.007649
|
|
VEGF signaling pathway
|
0.012929
|
|
B cell receptor signaling pathway
|
0.013733
|
|
ErbB signaling pathway
|
0.023767
|
|
mRNA surveillance pathway
|
0.02754
|
|
SNARE interactions in vesicular transport
|
0.002067
|
|
Non-small cell lung cancer
|
0.011991
|
|
Pathogenic Escherichia coli infection
|
0.012924
|
|
NOD-like receptor signaling pathway
|
0.016014
|
|
Bacterial invasion of epithelial cells
|
0.033175
|
|
Leishmaniasis
|
0.040738
|
|
African trypanosomiasis
|
0.014259
|
|
Vasopressin-regulated water reabsorption
|
0.025544
|
|
Endocrine and other factor-regulated calcium reabsorption
|
0.036159
|
|
Thyroid cancer
|
0.037167
|
Gene network analysis
We integrated different interactions in order to carry out gene network analysis, including enzyme-enzyme, protein-protein and gene expression interaction. Briefly, we downloaded data in KEGG database via KEGGSOAP (http://www.bioconductor.org/packages/2.4/bioc/html/KEGGSOAP.html) using R (http://www.r- project.org/) software, analyzed the interaction between gene regulation and protein modification. Enzyme-enzyme interaction indicated two enzymes catalyzing successive reaction steps. The data on protein-protein interaction was downloaded in MIPS database (http://mips.helmholtz-muenchen.de/proj/ppi/). The co-cition algorithm was implemented to analyze the gene expression interactions of literatures which had been reported. The higher frequency of co-citation gene was, the greater probability of gene expression interaction was. Finally, we integrated synthetically the gene network by the above three results which indicated the interaction among genes. Genes had high line connectivity in the network were deemed to hub gene which tended to play a central role in the stability of the network. Generally, most hub genes were transcription factors, sometimes, may be kinase. In this study, gene network analysis showed it’s a network diagram (Figure 3A) and a histogram (Figure 3B). The result revealed that SRRM1, HDAC1 and PRKED were hub genes.
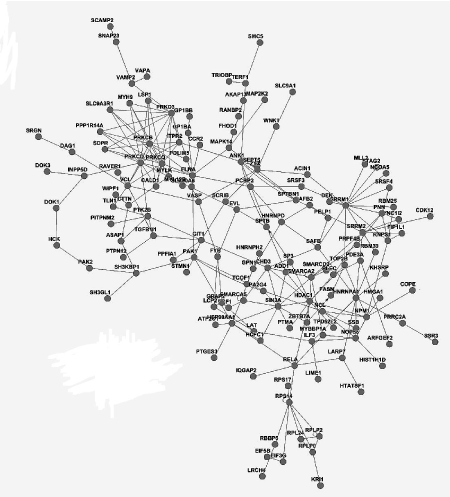
Figure 3A. Gene network analysis in PBMCs of MePGN, including the interaction of genes such as expression, binding and post-transcription modification
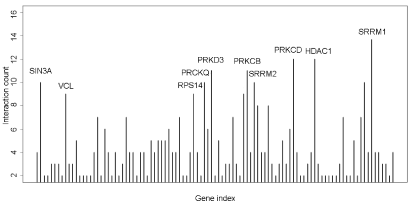
Figure 3B. Connectivity analysis in the gene network showed that SRRM1, HDAC1 and PRKCD were higher connectivity genes
The World Health Organization Committee have defined MePGN as an essentially uniform increase in mesangial cells in more than 80% of the glomeruli [13]. Cell proliferation and fibrosis are typical signs of it and may ultimately result in end-stage renal disease. Several recent reviews indicate that cytokines, histone H3, lysine 4, P2X7 receptors in PBMCs of chronic kidney disease play a critical roles in the process of paroxysm [14-16]. Thus, alteration of proteins in PBMCs of MePGN patients was considered to be related to the pathogenesis. Protein phosphorylation is an important post-translational modification to cell development, differentiation and signal transduction. In this sense, phosphoproteins are identified comprehensively and may facilitate to understand such processes and enable us to find significant therapeutic targets to prevent and repair the injury of renal.
Most phosphoproteins can be phosphorylated at serine, threonine and tyrosine residues in mammal. The relative abundance of phosphoproteins will be altered when protein kinase or phosphatase is activated or inhibited, resulting in abnormal cellular biological function or metabolic pathway. Utilizing TiO2 enrichment technology combined with LTQ-Orbitrap XL device, this research identified 693 differential phosphoproteins corresponding to 439 annotated genes in MePGN compared with normal control. GO analysis of these differential phosphorylation sites and genes showed that protein or nucleic acid binding took up the largest proportion of molecular function, which involved in nucleobase, nucleoside, nucleotide and nucleic acid metabolic process in the nucleus.
Mesangial cell proliferation is one of the typical characters in MePGN. Kenichi Suga [17] showed that Hic-5 as a hydrogen peroxide (H2O2)-inducible focal adhesion protein is involved in changes in the mesangial cell phenotype to produce abnormal extracellular matrix remodeling in MePGN. The immune pathogenesis of MePGN considered generally is immune complexes deposit, inflammation and abnormal secretion of cytokines. MAPKs is serine and threonine signal transducing enzymes that can be activated by phosphorylation in response to extracellular stimuli of cytokines [18]. The MAPK signaling pathway has important biological functions. Activation or inhibition of MAPK pathways have been indicated to be a potential pathogenesis in systemic lupus erythematosus [19] and rheumatoid arthritis [20], may be associated with inflammation. Cytokine-stimulated gene expression in airway myocytes can be mediated by Src through MAPK signaling pathway [21]. The view that differentially expressed and significantly up-regulated of neutrophil gelatinase-associated lipocalin receptor by IL-1β in mesangial cells of human glomerular disease via MAPK activation has been suggested [22]. In this study, pathway analysis showed that MAPK signaling pathway, Focal adhesion and Fc gamma R-mediated phagocytosis were modified significantly in MePGN pathogenesis. Notably, regulation of MAPK signaling pathway is abnormally activated in response to phosphorylation in PBMCs of MsPGN. This result suggested the activity or expression of phosphoproteins in this pathway above was altered, mediated by differential genes between MePGN and normal. We had faith in that MAPK signaling pathway may be available for treating MsPGN via appropriate regulation or interventions, which supplied an experimental groundwork for researching the pathogenesis and novel therapy of MsPGN. However, future more researches are essential to verify the accuracy of the result using western blot or other appropriate methods.
HDAC1 can be phosphorylated at Ser421 and Ser423 residues, which promotes its enzymatic activity and complex formation [23-25]. HDAC1 activity is a critical role in normal embryonic kidney gene expression, growth, and differentiation [26]. It is also critical for mediating proliferation of renal epithelial cells and renal interstitial fibroblasts, as well as HDAC1 induced in response to inflammation and fibrosis in tubulointerstitial injury [27-30]. In this study, network analysis showed that SRRM1, HDAC1 and PRKED were significant regulator in the network. This result suggested that HDAC1 played a potentially significant role in the phosphorylation of PBMCs, which was an experimentally theoretical supplement for exploring the pathogenesis of MsPGN.
In conclusion, MAPK signaling pathway was an important metabolic way, HDAC1 played an important role in the phosphorylation. It suggested that abnormal protein phosphorylation may contribute to the pathogenesis of MsPGN. Moreover, interventions of the activities of the involved gene and pathways may be able to prevent or retard the progressive renal injury of MsPGN. Nonetheless, this study was preliminary research for unraveling the modifications of phosphoproteome. Further validation step and investigations were required to elucidate the pathogenesis of MsPGN.
This work was financially supported by Science and Technology planning Project of Guangdong province (No.2016A020215029), Science and Technology Planning Project of Guangdong Province(No.2017B020209001), the Natural Science Foundation of Guangxi (No. 2017JJA130240), and Science research projects of Pingshan health and family planning system (No.201507), and we also sincerely thank the patients who participated in this study.
The present study was approved by the Ethics Committee of Guilin 181st hospital, which abides by the Helsinki Declaration on ethical principles for medical research involving human subjects.
We declare that we have no conflict of interest.
- Huzmeli C, Candan F, Seker A, Yildiz E, Terzi H, et al. (2016) C3 mesangial proliferative glomerulonephritis initially presenting with atypical hemolytic uremic syndrome: a case report. J Med Case Rep 10: 206. [Crossref]
- Yashima A, Mizuno M, Yuzawa Y, Shimada K (2017) Mesangial proliferative glomerulonephritis in murine malaria parasite, Plasmodium chabaudi AS, infected NC mice. Clin Exp Nephrol 21: 589-596. [Crossref]
- Qin D, Morita H, Inui K, Tayama H, Inoue Y, et al. (2013) Aldosterone mediates glomerular inflammation in experimental mesangial proliferative glomerulonephritis. J Nephrol 26: 199-206. [Crossref]
- Tan Z, Wang L, Wang C, Gao L, Yang Y (2015) Multicentric Castleman's disease with renal amyloidosis and mesangial proliferative glomerulonephritis: a case report. Int J Clin Exp Med 8: 2966-2973. [Crossref]
- Wang C, Feng Y, Wang M, Pi X, Tong H, et al. (2015) Volatile Organic Metabolites Identify Patients with Mesangial Proliferative Glomerulonephritis, IgA Nephropathy and Normal Controls. Sci Rep 5: 14744. [Crossref]
- Sun Y, Hu B, Fan C, Jia L, Zhang Y, et al. (2015) iTRAQ-based quantitative subcellular proteomic analysis of Avibirnavirus-infected cells. Electrophoresis 36: 1596-1611. [Crossref]
- Johansson HJ, Sanchez BC, Forshed J, Stål O, Fohlin H, et al. (2015) Proteomics profiling identify CAPS as a potential predictive marker of tamoxifen resistance in estrogen receptor positive breast cancer. Clin Proteomics 12: 8. [Crossref]
- Sui W, Tang D, Zou T, Zou G, Chen J, et al. (2013) Differential proteomic analysis of renal tissue in mesangial proliferative glomerulonephritis using iTRAQ technology. J Nephrol 26: 191-198.
- Lenton S, Grimaldo M, Roosen-Runge F, Schreiber F, Nylander T, et al. (2017) Effect of Phosphorylation on a Human-like Osteopontin Peptide. Biophys J 112: 1586-1596. [Crossref]
- Zhang X, Ma H, Huang J, Dai Y (2012) Characterization of the Phosphoproteome in SLE Patients. PLoS One 7: e53129. [Crossref]
- Bai B, Tan H, Peng J (2017) Quantitative Phosphoproteomic Analysis of Brain Tissues. Methods Mol Biol 1598: 199-211. [Crossref]
- Beltejar MG, Lau HT, Golkowski MG, Ong SE, Beavo JA (2017) Analyses of PDE-regulated phosphoproteomes reveal unique and specific cAMP-signaling modules in T cells. Beavo Proc Natl Acad Sci USA 114: 6240-6249.
- Arias LF, Taborda-Murillo A (2017) Mesangial proliferative glomerulonephritis: A glomerular disease or a non-specific morphological change? Nephrology (Carlton) 22: 575. [Crossref]
- Ditting T, Freisinger W, Rodionova K, Schatz J, Lale N, et al. (2016) Impaired excitability of renal afferent innervation after exposure to the inflammatory chemokine CXCL1. Am J Physiol Renal Physiol 310: 364-371.
- Zhang L, Dai Y, Wang L, Peng W, Zhang Y, et al. (2011) CpG array analysis of Histone H3 Lysine 4 Trimethylation in Peripheral Blood Mononuclear Cells of Uremia Patients. DNA and Cell Biology 30: 179-186.
- Lajdova I, Oksa A, Chorvat D, Topor P, Spustova V (2012) Purinergic P2X 7 receptors participate in disturbed intracellular calcium homeostasis in peripheral blood mononuclear cells of patients with chronic kidney disease. Kidney Blood Press Res 35: 48-57.
- Suga K, Kondo S, Matsuura S, Kinoshita Y, Urushihara M, et al. (2012) Glomerular expression of hydrogen peroxide-inducible clone-5 in human and rat progressive mesangial proliferative glomerulonephritis. Nephron Exp Nephrol 120: 59-68.
- Chen WP, Jin GJ, Xiong Y, Hu PF, Bao JP, et al. (2018) Rosmarinic acid down-regulates NO and PGE2 expression via MAPK pathway in rat chondrocytes. J Cell Mol Med 22: 346-353. [Crossref]
- Zhang F, Wu L, Qian J, Qu B, Xia S, et al. (2016) Identification of the long noncoding RNA NEAT1 as a novel inflammatory regulator acting through MAPK pathway in human lupus. J Autoimmun 75: 96-104.
- Singer CA, Lontay B, Unruh H, Halayko AJ, Gerthoffer WT (2011) Src mediates cytokine-stimulated gene expression in airway myocytes through ERK MAPK. Cell Commun Signa 9: 9-14.
- Jeong YC, Pyun K, Kwon YB (2014) Inhibition of mitogen-activated protein kinases phosphorylation plays an important role in the anti-nociceptive effect of pregabalin in zymosan-induced inflammatory pain model. Biol Pharm Bull 37: 1694-1698. [Crossref]
- Mao S, Jiang T, Shang G, Wu Z, Zhang N (2011) Increased expression of neutrophil gelatinase-associated lipocalin receptor by interleukin-1beta in human mesangial cells via MAPK/ERK activation. Int J Mol Med 27: 555-560.
- Nishihara K, Shahane SA, Xia M (2016) Determination of Histone H2AX Phosphorylation in DT40 Cells. Methods Mol Biol 1473: 71-76. [Crossref]
- Lai L, Cheng P, Yan M (2019) Aldosterone induces renal fibrosis by promoting HDAC1 expression, deacetylating H3K9 and inhibiting klotho transcription. Mol Med Rep 19: 1803-1808.
- Tang J, Yan Y, Zhao TC, Bayliss G, Bayliss G, et al. (2013) Class I histone deacetylase activity is required for proliferation of renal epithelial cells. Am J Physiol Renal Physiol 305: 244-254.
- Kiweler N, Brill B, Wirth M, Breuksch I (2018) The histone deacetylases HDAC1 and HDAC2 are required for the growth and survival of renal carcinoma cells. Arch Toxicol 92: 2227-2243. [Crossref]
- Chen S, Bellew C, Yao X, Stefkova J, Dipp S, et al. (2011) Histone deacetylase (HDAC) activity is critical for embryonic kidney gene expression, growth, and differentiation. J Biol Chem 286: 32775-32789.
- Li H, Wang X, Zhang C, Cheng Y, Yu M, et al. (2018) HDAC1-induced epigenetic silencing of ASPP2 promotes cell motility, tumour growth and drug resistance in renal cell carcinoma. Cancer Lett 432: 121-131. [Crossref]
- Choi SY, Kee HJ, Kurz T (2016) Histone deacetylase inhibitors attenuate P-aIgA1-induced cell proliferation and extracellular matrix synthesis in human renal mesangial cells in vitro. Acta Pharmacol Sin 37: 228-34.
- Liu H, Chen S, Yao X (2018) Development. Histone deacetylases 1 and 2 regulate the transcriptional programs of nephron progenitors and renal vesicles. Development 145: 1-11.