High morbidity and mortality associated with oral cancers and its high incidence and prevalence necessitates earlier diagnosis and effective management. Despite of ease in morphological accessibility, oral cancers are still diagnosed mostly in advanced stages chiefly due to lack of effective and cheap screening tools.The search of a suitable biomarker, which can diagnose, oral cancer effectively and can predict the progression of Oral Potentially malignant Disorders (OMPD) to Oral Cancer with accuracy, still exists. PRAME (Preferentially Expressed Antigen of Melanoma) is one such biomarker, which is a dominant repressor of Vitamin A (Vit A). Vit A, chiefly Retinoic Acid (RA) is extensively used these days for chemoprevention owing to its role in cell growth and differentiation. RA emerged as a cheap and acceptable chemo preventive agent since RA is readily available in the form of Vit A food supplements, Vit A is being used in patients with various grades of OPMDs and oral cancer, but its efficiency is still debatable owing to its mixed results in various cancers. Inconsistency in efficient results led to study of various molecules like PRAME, which is involved in RA metabolic pathway thereby modulating the outcome and efficacy of Retinoic Acid chemoprevention.
Oral premalignant disorders, oral cancer, PRAME, Retinoic Acid
Abbreviations
PRAME: Preferrentially Expressed Antigen of Melanoma; OMPD: Oral Potentially malignant Disorders; RA: Retinoic Acid
Cancer of lip and oral cavity, according to GLOBOCAN 2018, stands 18th and 14th most common cancer worldwide according to incidence and mortality respectively, with an estimated 354,864 (2.0 %) new cases diagnosed leading to 177,384 (1.9%) number of deaths in 2018. South central Asia procures 2nd highest place for incidence of lip and oral cavity cancer (17.4%) [1].
Owing to high morbidity and mortality, oral cancers have become a grave problem and its high incidence and prevalence necessitates earlier diagnosis and effective management. Despite of ease in morphological accessibility, oral cancers are still diagnosed mostly in advanced stages chiefly due to lack of effective and cheap screening tools. Molecular/ genetic studies not only augment clinical assessment and classification of oral lesions but also predict malignant potential of oral lesions, thus reducing its incidence and increasing the scope for early diagnosis and treatment of oral cancers. The search of a suitable biomarker still exists which can diagnose oral cancer effectively and can predict the progression of OMPD to Oral Cancer with accuracy. PRAME is one such biomarker, which interacts with Retinoic Acid receptors thereby modulating the outcome and efficacy of Retinoic Acid chemoprevention. Herein we aim to review PRAME pathway, its role in oral potentially malignant disorders and oral cancers via its interaction with Retinoic acid.
Method of data collection
A Pubmed database search was performed using key words PRAME or Retinoic Acid and Oral Cancer. The search yielded about 848 articles. All the publications which included areas other than oral cavity were filtered using terms not melanoma, not leukaemia, not breast carcinoma, not liposarcoma, not medulloblastoma, not hepatocellular carcinoma, not lung carcinoma, not neuroblastoma, not ovarian cancer etc. Only human studies were included. After this filtration, 348 publications remained. All the publications were then manually screened by including only oral cancer, precancer and head and neck cancer studies and only those publications with atleast 2 citations and impact factor of atleast 1 were considered for review. Finally we were left with 26 studies, which were included in this review.
The social burden of cancer
Cancer is a generic term for a large group of diseases that can affect any part of the body. Though ‘cancer’ is not a medical term but widely used and accepted synonymously with neoplasia/ malignancy/ tumors. Certain characteristics are common but few distinctive features aid in differentiating these terms. However due to wide acceptance of the term cancer, herein we will be using the same to describe malignancy. It is noteworthy that term neoplasia includes both tumors (benign growths) and malignancies. All tumors do not progress to malignancy similarly all malignancies are not preceeded by tumors. Difference between malignancy and tumor are described briefly in Table 1.
Table 1. Table describing difference between a malignancy and tumor
Malignancy |
Tumor |
Rapid growth due to uncoordinated and uncontrolled cell division |
Rapid growth due to uncontrolled cell growth which may /may not be uncoordinated |
Growth beyond boundaries to invade adjacent or distant tissues (metastasis present) |
Growth restricted to the tissue of origin (metastasis absent) |
Continued growth even after removal of causative stimuli |
Further growth stops once causative stimuli is removed |
Commonly referred as cancer |
Commonly referred as benign tumors |
The GLOBOCAN 2018 database reports that the global cancer burden is estimated to have risen to 18.1 million new cases and 9.6 million deaths in 2018 as compared to the 8.8 million deaths reported in 2015. WHO projected its rise by 70% till 2030. One in 5 men and one in 6 women worldwide develop cancer during their lifetime, and one in 8 men and one in 11 women die from the disease. Worldwide, the 5-year prevalence (total number of people who are alive within 5 years of a cancer diagnosis) is estimated to be 43.8 million.
It was observed that approx. 70% deaths from cancer occur in middle- and lower-income countries. However, a shift is observed from cancers related to poverty and infections to cancers associated with lifestyles more typical of industrialized countries. Global patterns show that for men and women combined, nearly half of the new cases and more than half of the cancer deaths worldwide in 2018 are estimated to occur in Asia [1].
Oral cancer and oral pre cancer
To define anatomically, oral cancer is a malignant neoplasia arising on lips, the lining inside the cheeks and lips, the anterior two thirds of the tongue, the upper and lower gingiva, the floor of the mouth under the tongue, the roof of the mouth, and the small area behind the third molar [2].
Oral cancer is the 2nd most common cancer in India amongst men (11.28% of all cancers), 5th most frequently occurring cancer amongst women (4.3% of all cancers) and the 3rd most frequently occurring cancer in India amongst both men and women [3].
It has been well-described in the literature that virtually all oral cancers are preceded by visible clinical changes in the oral mucosa usually in the form of white or red patch (two-step process of cancer development).
WHO in 1978 used the term ‘precancer’ which was further classified into ‘lesions’ and ‘conditions’
- A precancerous lesion is “a morphologically altered tissue in which oral cancer is more likely to occur than its apparently normal counterpart.” These precancerous lesions include leukoplakia, erythroplakia, and the palatal lesions of reverse smokers.
- A precancerous condition is “a generalized state associated with significantly increased risk of cancer.” The precancerous conditions include submucous fibrosis, lichen planus, epidermolysisbullosa, and discoid lupus erythematous.
However, the use of the term OPMDs (oral potentially malignant Disorder) and elimination of the term ‘precancer’ was recommended in a workshop held in London 2005, owing to the findings that not all precancers progress to cancers [4].
Earlier it was considered that origin of a malignancy in the oral cavity would correspond with the site of precancerous lesion. On the other hand, in precancerous condition, cancer may arise in any anatomical site of the mouth or pharynx. Today we know that even the clinically ‘normal’ appearing mucosa in a patient with precancerous lesion may have dysplasia on the contralateral anatomic site or molecular aberrations in other oral mucosal sites suggestive of a pathway to malignant transformation and that cancer could subsequently arise in apparently normal tissue.
Biomarkers in carcinogenesis: need
Cancer development (carcinogenesis) is a complex multistep process wherein a stimulus (carcinogen) incurs either genetic or epigenetic changes that enable the cell to achieve certain characteristic carcinogenic features referred to as hallmarks of cancer, which are acquired functional capabilities that allow cancer cells to survive, proliferate, and disseminate. These characteristics are acquired in different tumor types via distinct mechanisms and at various times during the course of multistep tumorigenesis /carcinogenesis [5]. Briefly these characteristics/ hallmarks are
- Sustaining growth potential/proliferative signaling
- Evading growth suppressors/insensitivity to growth suppressors
- Resisting apoptosis/evasion of apoptosis
- Limitless replicative potential
- Inducing sustained angiogenesis
- Tissue invasion and metastasis
- Reprogramming of cellular energy metabolism
- Evading immune destruction
To confer malignant cells with capability to achieve above-mentioned hallmarks, many proteins/ molecules orchestrate in a definite manner. These proteins are technically defined by The National Cancer Institute as biomarkers / biological molecules found in blood, other body fluids or tissues representing a sign of a normal or abnormal process or condition of a disease like cancer [6].
Several molecular and technological markers are being explored in order to identify key biological molecules linked to cancer development, risk assessment, screening, recurrence prediction, indicating prognosis, invasion/ metastasis and monitoring therapeutic responses of cancer.
To date, no biomarkers predicting favorable outcome or response to definitive cancer treatment are available. Most of the markers, such as tumor size or extracapsular spread, appear to be inadequate for the risk stratification of patients with oral cancer, for predicting recurrence and for predicting malignant transformation of precancer to cancer. There is a growing need for clinically - relevant and practical biomarkers that will allow the appropriate selection of patients who might benefit from desired cancer treatment strategies.
A category of biomarkers known as Tumor Antigens (TAs), have emerged as potentially important targets for antigen-specific cancer immunotherapy. Tumor antigens (TAs) are tumor proteins that, when expressed in tumors, are recognized by the host immune system. They represent markers that are either specific for individual tumors or are generally overexpressed in tumors as compared to normal tissues [7].
TAs can be neoantigens (tumor-specific antigens/ TSAs) that arise from mutation or RNA splicing and are expressed only by cancer cells and not by normal tissue or Tumor-associated antigens/ TAAs show increase in expression in cancer tissue as compared to normal tissue [8].
Conceptually, tumor antigens can be divided into different groups based on their expression patterns and the presence of underlying gene mutations. Firstly, they may be specific products of malignant cells due to mutations, translocations, and other distinct genetic events (e.g., the BCR/ABL fusion protein in leukemia). Secondly tumor cells may express the wild-type version of proto-oncogenes at a higher level than their normal cellular counterparts (e.g. as a result of amplification as for HER-2/neu in breast cancer) and thirdly a class of nonmutated genes whose expression, with exception of testis and fetal tissues, seems to be mostly restricted to tumor cells e.g. cancer-testis antigens (CTA) include the MAGE, GAGE/PAGE, BAGE, LAGE/NY-ESO-1, and PRAME.
PRAME: Novel biomarker in oral carcinogenesis
Preferentially expressed antigen of melanoma (PRAME) was first detected as a tumour related/ associated antigen in cells isolated from a melanoma, and high PRAME expression had been detected in 88–95% of primary melanomas [9].
According to Entrez Gene, PRAME is also referred to as MAPE (melanoma antigen preferentially expressed in tumors), OIP4 or CT130 (opa-interacting protein 4 0r cancer/testis antigen 130) and LOC23532.
PRAME gene encodes a 509 Amino acid protein (Mol. Mass 57890 Da), located on the reverse strand of human chromosome 22 (22q11.22). It covers 12.06 kb, from 22902176 to 22890113 and has 8 exons and 15 distinct GT-AG introns. It is located within the human immunoglobulin lambda locus which contains a large number of V1 gene segments for light chain production during B cell development [10].
PRAME is not expressed or only slightly expressed in normal tissues, except testicles, ovaries, endometrium and adrenal glands. However, high levels of PRAME are reported in malignant cells, such as acute and chronic leukemia, primary and metastatic melanoma, Hodgkin’s lymphoma, breast cancer, and head and neck squamous cell carcinoma [11-16].
Functionally, the gene has been observed to be associated to diseases (Acute Disease; Chromosome Aberrations; Kidney Neoplasms; T-Cell Leukemia; Non-Hodgkin Lymphoma; Melanoma; Multiple myeloma; Residual and Recurrence Neoplasm). The various functions which PRAME is proposed to participate in are schematically represented in figure 1 a & b [17].
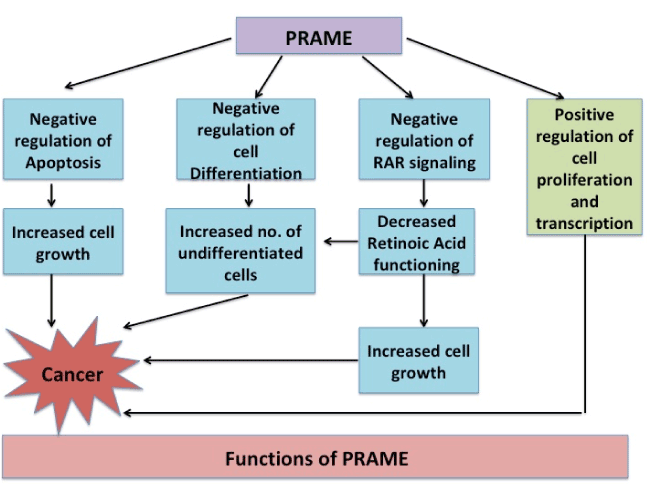
Figure 1(a). PRAME not only negatively regulates cell apoptosis, differentiation and RAR signaling but also positively regulates cell growth leading to increased cell growth and ultimately to Cancer
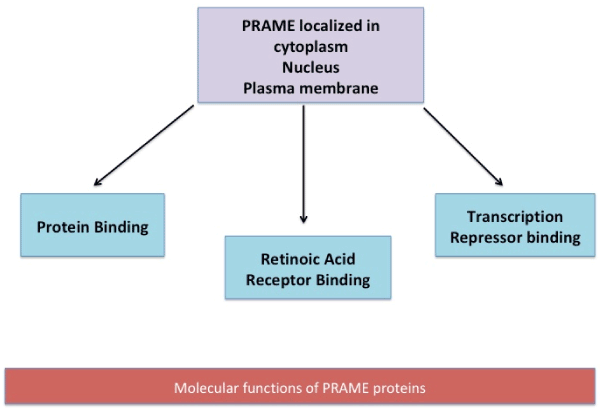
Figure 1(b). At cellular levels, PRAME is located in the cytoplasm, nucleus and plasma membrane. PRAME protein aids in transcription repressor binding, Retinoic Acid receptor binding and protein binding
Since PRAME is a tumor associated antigen, it is recognized by autologous cytotoxic T cells against surface antigens. PRAME expression is retained in the presence of antitumor T cell responses, suggesting that expression of PRAME confers to tumor cells a selective advantage that outweighs the CTL-mediated tumor cell killing. This antigen is expressed in varying cancer types and its expression in tumor cells has an impact on prognosis and survival of cancer patients. In most cases, high expression of PRAME is a marker for poor prognosis, increased development of metastasis and low disease-free survival [18-20].
The cause of aberrant PRAME expression in malignancies is largely unknown. Because many studies have relied on mRNA-based techniques, it is clear that PRAME transcript levels are very highly induced in tumor tissues, but whether this is due to gene amplifications, enhanced transcription rates, or altered mRNA turnover has not been addressed. However, genetic alterations, such as mutations or translocations in PRAME, have not been reported [21].
EPPING MT (2005) indicated that PRAME may be instrumental in disease progression by interfering with retinoic acid (RA) receptor (RAR) signaling. PRAME is a dominant repressor of RAR signaling: upon binding to RAR in the presence of RA, PRAME prevents ligand-induced receptor activation, antagonizing RAR signaling. Moreover, the restoration of sensitivity to RA in RA-resistant cells can be obtained by the knockdown of PRAME by RNA interference [22,23]. RA signalling is essential in development, cell fate determination, and tissue homeostasis. RA induces transcription of a set of target genes by binding to and activation of its receptor, resulting in differentiation and cell cycle arrest in responsive cells [24].
In normal cells, cell cycle arrest, differentiation or apoptosis does not occur as target genes are not activated due to binding of repressor complex to the retinoic acid receptor (RAR) on RA-Responsive Element (RARE) When Retinoic Acid binds to coactivator complex and RAR on RARE it acts upon target gene to cause cell cycle arrest, differentiation and apoptosis. In presence of PRAME, which replaces the coactivator complex, RA binds to RAR on RARE thus blocking its action on target genes and thus causing suspension of cell cycle arrest, differentiation and apoptosis therefore supporting cancer formation [22]. (Figure 2a and 2b).
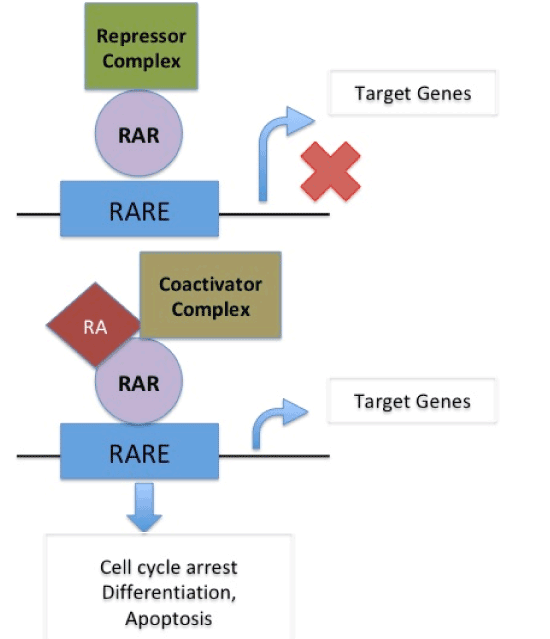
Figure 2 (a). In normal cells, cell cycle arrest, differentiation or apoptosis does not occur as target genes are not activated due to binding of repressor complex to the retinoic acid receptor (RAR) on RA-Responsive Element (RARE). When Retinoic Acid binds to coactivator complex and RAR on RARE it acts upon target gene to cause cell cycle arrest, differentiation and apoptosis
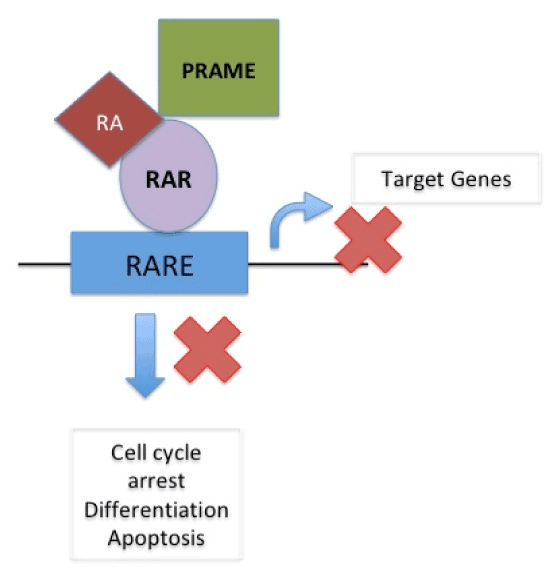
Figure 2 (b). In presence of PRAME, which replaces the coactivator complex, RA binds to RAR on RARE thus blocking its action on target genes and thus causing suspension of cell cycle arrest, differentiation and apoptosis therefore supporting cancer formation. (EPPING MT, WANG L, EDEL MJ, CARLÉE L, HERNANDEZ M, BERNARDS R. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signalling
The binding of Retinoic acid with cellular RA binding protein (CRABP) allows it to enter the nucleus where it can bind, and serve as ligand, with various nuclear receptors including retinoic acid receptor (RAR) and retinoic X receptor (RXR), to mediate genomic and non-genomic mechanisms [25].
Binding of the retinoic acid ligand to RAR causes conformational changes that modulate receptor complex function. In addition, these receptor complexes have a range of additional co-activators and co-repressors that modulate receptor activity. Retinoids exert most of their effects by modulating gene expression [26].
Retinoids, the derivatives of vitamin A, are the most-studied chemopreventive agents, and 13-cis-retinoic acid is the best-studied chemopreventive retinoid.
Importance of vitamin A pathway
Oral cancer chiefly involves non- keratinized stratified squamous epithelium. It was observed that these nonkeratinizing squamous cells can undergo keratinization in vivo during vitamin A deficiency, indicating that the maintenance of the nonkeratinizing state of squamous cells depends on the continuous presence of Retinoic acid/ Vitamin A. Retinoids exert profound effects on the growth and differentiation of normal and malignant epithelial cells, also Vitamin A and its analogues delay tumour appearance, retard tumour growth and regress tumours [27-29].
It was observed that retinoids suppress the proliferation of head and neck cancer cells in monolayer cultures and inhibit the formation of colonies in semisolid agarose and decrease the growth of multicellular spheriods. In addition, retinoids also suppress the differentiation markers K1 keratin, type 1 transglutaminases and involucrin [30-34].
Several studies have been attempted with various study groups like squamous cell carcinomas of tongue in rats and hamsters, normal oral keratinocyte and neoplastic cell lines revealing that Retinoic Acid greatly suppresses development and progression of Squamous cell carcinoma [35-37].
Some of the important studies and their observation are listed as Table 2.
Table 2. Various studies done on Retinoic acid, oral cancers and precancers
S No. |
Observations |
Inference |
1 |
Presence of significantly higher levels of CRBP (cellular Retinol Binding Protein) and CRABP (cellular Retinoic Acid Binding Protein) in epidermoid carcinomas of oral cavity and oropharynx as compared to adjacent normal tissue. |
Retinol, retinoic acid and their analogs inhibit development of epithelial tumors mediated by binding of these proteins to their receptors |
2 |
Growth inhibitory effect of 9 –Cis- Retinoic Acid (9CRA) on oral squamous cell carcinoma depends upon the expression levels of RARs, especially RAR beta proteins and RXR alpha proteins. thus suggesting. |
9 CRA may be a powerful therapeutic agent for head and neck cancers |
3 |
The role of RARβ in growth and differentiation in two oral squamous cell lines HSC-4 and Ho-1-N-1 established from a well-differentiated squamous cell carcinoma of the tongue and cheek respectively was studied.
|
Both the cell lines displayed growth inhibition and underwent morphological changes. RARβ-transfected HSC-4 clones underwent apoptosis even in the absence of 9CRA treatment. In contrast, RARβ-transfected Ho-1-N-1 clones exhibited cell cycle arrest without undergoing apoptosis initially; however, apoptosis was induced in these cells after 6 days of 9CRA treatment. |
4 |
The effect of retinoids on the invasive potentials of oral SCC cells in vitro was investigated, and it was demonstrated that β- All Trans Retinoic Acid (ATRA) and 13-cisRetinoic Acid (CRA) inhibited the growth of the oral SCC cells (BHY and HNt) in vitro, but low-dose ATRA and CRA marginally affected the growth of the cells. Furthermore, low-dose RAs enhanced the in vitro invasiveness of BHY cells. |
If the intra- tumoral concentration of RAs does not reach the antiproliferative dose, RAs might enhance the invasion and migration of the cancer cells. Then RAs should be used at enough high concentration to show the anti-proliferative effect or differentiation-inducing effect on the cancer cells. |
5 |
The Toxicity-to-risk balance of (13 CRA) was observed to be delicate and complicated. 13 CRA, at high doses, has an established chemo preventive activity involving suppressing oral premalignancy and preventing second primary tumours. However high-dose 13 CRA is not ideal for widespread chemoprevention approaches because of its toxicity. |
Careful calculation of dose to risk ratio needs to be evaluated to outweigh the efficacy of 13 CRA |
6 |
Retinoic acid receptor-β2 (RAR-β2) expression is observed to be suppressed in oral premalignant lesions and head and neck squamous cell carcinomas. RAR-β2 gene expression in such lesions can be silenced by promoter methylation. |
5-Aza-CdR can restore RAR-β2 inducibility by ATRA in most cell lines, and the combination of 5-aza-CdR and ATRA is more effective in growth inhibition than single agents. |
7 |
Expression of the three retinoic acid receptor (RAR) (alpha, beta, gamma) mRNAs in cell lines cultured from oral squamous cell carcinoma and from benign, hyperplastic, and hyperkeratotic (leukoplakia) lesions arising in various regions of the oral cavity were observed. It was also observed that nuclear retinoic acid receptor beta (RAR-beta) expression decreases in human premalignant oral lesions |
Abnormally low expression of the RAR beta receptor contributed to neoplastic progression in stratified squamous epithelia and may help to determine whether a tumor is responsive to RA as a chemotherapeutic agent. |
8 |
Significant increase in RARα immunopositivity in oral SCCs compared to normal tissue and hyperplastic lesions was observed. One intriguing feature was the significant decrease in RARβ immunopositivity in hyperplastic lesions compared with normal oral mucosa as well as in oral SCCs compared with normal tissues. |
RARα immunopositivity can be used to predict establishment and progression of Oral SCC from normal tissue via hyperplastic lesions. |
In order to use retinoids more effectively on the patients with oral SCC, the effect of retinoids not only on the proliferation of the cells but also on the other biological characteristics, such as invasiveness or angiogenesis, should be examined.
The utilization of retinoids in head and neck squamous cell carcinoma chemoprevention has a long history.
Several potential chemopreventive agents have been evaluated including vitamin A (retinylpalmitate), other retinoids, selenium, vitamin E and COX-2 inhibitor [38].
HONG WK et al (1986) gave 13-cis-retinoic acid, 1 to 2 mg per kilogram of body weight per day for three months, and followed them for six months post treatment and reported a decrease in 67 % patients, dysplasia was reversed in 54 percent and in 57 % patients the clinical response to the drug correlated with the histologic response [39].
A randomized trial involving tobacco users with oral leukoplakia supplemented with 200,000 IU of vitamin A every week for 6 months has shown complete remissions in 57% and a progression arrest in 100% of the treated group as opposed to 3 and 21% in the placebo group, respectively [40].
In another study, complete or partial remissions of premalignant lesions have been observed in 45% of patients treated with one of three different retinoids after a 6-year follow-up. Unfortunately, this therapy had considerable toxicity [41].
Although results of clinical trials with vitamin A or retinoids in oral leukoplakia or premalignancy were cautiously quite promising, there were several cases where Vitamin A or its derivatives could not yield satisfactory results. Also, retinoids did not reliably improve the outcome in head and neck cancers (HNSCC). For example, in a Phase III study involving 103 patients with stage I–IV HNSCC, who received either 50–100 mg/m2 isotretinoin daily or a placebo, no significant differences in the frequency of secondary primary tumors or in the number of local, regional or distant recurrences of the primary cancers were observed [42].
RHEE JJ et al also reported that despite the prominent RAR-β expression in human HNSCC and in contrast to patients with premalignant lesions, retinoid therapies were largely ineffective in patients with HNSCC. Although low-dose retinoids showed limited therapeutic effectiveness, significant toxicity of high- dose retinoids has further hindered their use in HNSCC therapy [38].
These observations encouraged investigators to realise the mechanistic underpinning of such therapeutic responses. In view of prominent PRAME overexpression in HNSCC, the lack of therapeutic efficacy with retinoids is not surprising.
As described by EPPING MT et al (2005) PRAME interferes with retinoic acid (RA) receptor (RAR) signalling which is known to regulate various aspects of cell proliferation, differentiation, apoptosis and vertebrate development and may be thus instrumental in cancer progression [21,22].
Hence failure to recognize PRAME as a factor implicated in RA resistance led to such ambiguous results of the various previous clinical trials evaluating the usefulness of retinoids in chemoprevention. Therefore, selection of PRAME-negative patients for RA trials could result in better clinical responses.
PRAME in oral precancer and cancer: its importance
Hence failure to recognize PRAME as a factor implicated in RA resistance led to such ambiguous results of the various previous clinical trials evaluating the usefulness of retinoids in chemoprevention. Therefore, selection of PRAME-negative patients for RA trials could result in better clinical responses.
As described previously high levels of PRAME are reported in malignant cells, such as acute and chronic leukemia, primary and metastatic melanoma, Hodgkin’s lymphoma, breast cancer, and head and neck squamous cell carcinoma etc. evaluated by semiquantitative (RT-PCR) expression of PRAME gene in surgical samples of the tumors, margins, and lymph nodes (when present) from patients with a diagnosis of head and neck carcinoma and observed a variable intensity of expression with respect to tumor staging, and smoking habits. Although it was observed that PRAME gene was always present in the metastatic lymph node [43].
It has been suggested that PRAME contributes to cancer development and progression by interfering with the metabolic pathway of all-trans retinol (vitamin A) and its active metabolites, retinal, β-carotene, all-trans retinoic acid (ATRA), and 9-cis and 13-cis retinoic acids, collectively known as retinoids.
Since PRAME is a dominant repressor of RAR signalling. It prevents ligand-induced receptor activation upon binding to RAR in the presence of RA. Thus, cancer cells over expressing PRAME acquire a survival advantage and are able to escape from RA-induced cell growth arrest. Thus, PRAME confers growth or survival advantages and promotes malignant differentiation of stem cells [44].
Therefore, malignant and precancerous cells are benefitted via loss of RA responsiveness associated with PRAME overexpression. Also the presence of the PRAME in oral cancer cells could explain a lack of the past success in chemoprevention with retinoids in patients with pre-cancerous oral lesions [45,46].
SZCZEPANSKI MJ et al (2013) studied HNSCC cell lines and demonstrated that PRAME protein and mRNA were overexpressed in tumor cells but not in normal keratinocytes (HaCaT cells).
They also evaluated PRAME expression in precancerous dysplastic lesions of the head and neck obtained from 12 patients. Only 8/12 patients (66%) were positive for PRAME.
Also, PRAME silencing with siRNA in HNSCC cells followed by co incubation with clinically relevant concentrations of RA decreased in vitro migration of these cells and induced their apoptosis.
PRAME overexpression in HNSCC tissue specimens, as determined by immunohistochemistry, also correlated with the conventional markers of poor prognosis such as a large tumor size, high tumor grade and lymph node involvement.
Furthermore, PRAME was found to be overexpressed in HNSCC of patients with advanced disease (stages III and IV).
In aggregate, these data suggested that elevated PRAME expression in HNSCC could potentially serve as a biomarker of poor outcome and as a future therapeutic target [47].
PRAME: a future perspective
Lack of therapeutic inefficiency of vitamin A/ Retinoids lead to search of different therapeutic strategies targeting PRAME.
Preclinical studies aimed at decreasing PRAME expression in cancer cell lines by delivery of PRAME-specific siRNA resulted in a cell cycle arrest and apoptosis [48].
Also, PRAME is immunogenic and induces cytolytic T-lymphotoxicity hence can serve as target antigens in antitumor vaccination trials. Although PRAME-based vaccines have not been so far tested in clinical trials, preclinical studies confirm that PRAME can induce cytolytic T lymphocytes with antitumor activity [49].
Given that PRAME is frequently and selectively overexpressed in HNSCC, its utilization as an effective immunogen in antitumor vaccines or, alternatively, adoptive transfers of PRAME-specific T cells, might result in positive immunologic and possibly also clinical responses, especially when immunotherapy is followed by retinoid-based therapy [50].
Therefore, it might be expected that immunotherapies targeting PRAME and delivered in the form of peptides or proteins pulsed on antigen-presenting cells plus an adjuvant or retinoid might become an efficacious therapeutic modality for HNSCC patients in the future.
High mortality and morbidity associated with oral cancers necessitates its early diagnosis and effective management. PRAME is emerging as a biomarker of outcome in various cancers and precancers. Retinoic Acid and its derivatives have been observed to aid in chemoprevention due to its ability to control cell growth, prevent keratinisation and promoting apoptosis. However inconsistent results observed in various studies required a detailed insight into the underlying metabolic mechanism. Evidence for PRAME overexpression in oral cancers and its dominance in repression of Retinoic acid binding to its receptor has provided a partial explanation for the paucity of therapeutic successes with retinoids in the past.
The future therapeutic strategies should be based on targeting PRAME by combination therapies with siRNA and retinoids.
In chemoprevention, selection of patients with PRAME-negative premalignant lesions for retinoid-based preventive treatments might result in better clinical responses.Considering the role of PRAME in the RA metabolism, it is suggested that only patients with the premalignant lesions negative for PRAME are likely to be responsive to retinoid therapy and may be the best potential candidates for chemoprevention with retinoids.
Authors are thankful to DHR-MRU facility at KGMU, Lucknow
None
Authors declare that there are no competing interests.
None
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, et al. (2018) Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries CA. Cancer J Clin 68: 394-424.
- No authors listed (2015) Abstracts from the 38th annual meeting of the society of general internal medicine. J Gen Intern Med 30 Suppl 2: 45-551. [Crossref]
- https;//www.Cancerindia.org.in/statistics
- Warnakulasuriya S, Johnson NW, van der Waal I (2007) Nomenclature and classification of potentially malignant disorders of the oral mucosa. J Oral Pathol Med 36: 575-580. [Crossref]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646-674. [Crossref]
- Mishra A, Verma M (2010) Cancer biomarkers: are we ready for the prime time? Cancers (Basel) 2: 190-208. [Crossref]
- HaenSP, Rammensee HG (2013) The repertoire of human tumor-associated epitopes--identification and selection of antigens and their application in clinical trials. Curr Opin Immunol 25: 277-283.
- Van Den Eynde BJ, Van Der Bruggen P (1997) T cell defined tumor antigens. Curr Opin Immunol 9: 684-693.
- Ikeda H, Lethe B, Lehmann F, Van Baren N, Baurain JF, et al. (1997) Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 6: 199-208.
- Kawasaki K, Minoshima S, Nakato E, Shibuya K, Shintani A, et al. (1997) One-megabase sequence analysis of the human immunoglobulin lambda gene locus. Genome Res 7: 250-261.
- Schenk T, Stengel S, Goellner S, Steinbach D, Saluz HP (2007) Hypomethylation of PRAME is responsible for its aberrant overexpression in human malignancies. Genes Chromosomes Cancer 46: 796-804.
- Epping MT, Hart AA, Glas AM, Krijgsman O, Bernards R (2008) PRAME expression and clinical outcome of breast cancer. Br J Cancer 99: 398-403.
- Tan P, Zou C, Yong B, Han J, Zhang L, et al. (2012) Expression and prognostic relevance of PRAME in primary osteosarcoma. Biochem Biophys Res Commun 419: 801-808.
- Oberthuer A, Hero B, Spitz R, Berthold F, Fischer M (2004) The tumor-associated antigen PRAME is universally expressed in high-stage neuroblastoma and associated with poor outcome. Clin Cancer Res 10: 4307-4313.
- Proto-Siqueira R, Falcao RP, De Souza CA, Ismael SJ, Zago MA (2003) The expression of PRAME in chronic lymphoproliferative disorders. Leuk Res 27: 393-396.
- Paydas S, Tanriverdi K, Yavuz S, Seydaoglu G (2007) PRAME mRNA levels in cases with chronic leukemia: Clinical importance and review of the literature. Leuk Res 31: 365-369.
- Tanaka N, Wang YH, Shiseki M, Takanashi M, Motoji T (2011) Inhibition of PRAME expression causes cell cycle arrest and apoptosis in leukemic cells. Leuk Res 35: 1219-1225.
- Yan M, Himoudi N, Basu BP, Wallace R, Poon E, et al. (2011) Increased PRAME antigen-specific killing of malignant cell lines by low avidity CTL clones, following treatment with 5-Aza-2'-Deoxycytidine. Cancer Immunol Immunother 60: 1243-1255.
- Van't Veer LJ, Dai H, Van De, Vijver MJ, He YD, et al. (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415: 530-536.
- Kewitz S, Staege MS (2013) Knock-down of PRAME increases retinoic acid signalling and cytotoxic drug sensitivity of Hodgkin lymphoma cells. PLoS One 8: e55897.
- Epping MT, Bernards R (2006) A causal role for the human tumor antigen preferentially expressed antigen of melanoma in cancer. Cancer Res 66: 10639-10642.
- Epping MT, Wang L, Edel MJ, Carlee L, Hernandez M, et al. (2005) The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signalling. Cell 122: 835-847.
- Gudas LJ, Wagner JA (2011) Retinoids regulate stem cell differentiation. J Cell Physiol 226: 322-330.
- Altucci L, Gronemeyer H (2001) The promise of retinoids to fight against cancer. Loss of RA responsiveness is therefore beneficial to cancer cells. Nat Rev Cancer 1: 181-193.
- Napoli JL (1996) Retinoic acid biosynthesis and metabolism. FASEB J 10: 993-1001.
- Boylan JF, Gudas LJ (1992) The level of CRABP-I expression influences the amounts and types of all-trans-retinoic acid metabolites in F9 teratocarcinoma stem cells. J Biol Chem 267: 21486-21491.
- Basu TK (1979) Vitamin A and cancer of epithelial origin. J Hum Nutr 33: 24-31.
- Lotan R (1993) Retinoids and squamous cell differentiation. In: W.K. HONG and R. LOTAN (eds.), Retinoids in Oncology 43-72.
- Jetten AM, Nervi C, Vollberg TM (1992) Control of squamous differentiation in tracheobronchial and epidermai epitheliai cells: role of retinoids. JNCI Monogr 13: 93-100.
- Relax M, Pitman SW, Sartorelli AC (1985) Modulation of the terminal differentiation of human squamous carcinoma cells in vitro by all-trans-retinoic acid. J Natl Cancer Inst 74: 1015-1023.
- Lotan R, Sacks PG, Lotan D, Hong WK (1987) Differential effects of retinoic acid on the in vitro growth and cell-surface glycoconjugates of 2 human head and neck squamous-cell carcinomas. Int J Cancer 40: 224-229.
- Jetten AM, Kim JS, Sacks PG, Rearick JI, Lotan D, et al. (1990) Suppression of growth and squamous cell differentiation markers in cultured human head and neck squamous carcinoma cells by ß-all trans retinoic acid. Int J Cancer 45: 195-202.
- Lotan R, Lotan D, Sacks PG (1990) Inhibition of tumor cell growth by retinoids. Methods EnzymoL 190: 100-110.
- Sacks PG, Oke V, Vasey T, Lotan R (1989) Modulation of growth, differentiation, and glycoprotein synthesis by ß-all-transretinoic acid in a multicellular tumor spheroid model for squamous carcinoma of the head and neck. Int J Cancer 44: 926-33.
- Huang CC (1986) Effect of retinoids on the growth of squamous cell carcinoma of the palate in rats. Am J Otolaryngol 7: 55-57.
- Goodwin WJ Jr, Bordash GD, Huijing F, Altman N (1986) Inhibition of hamster tongue carcinogenesis by selenium and retinoic acid. Ann Otol Rhinol Laryngol 95: 162-166.
- Rubin AL, Rice RH (1986) Differential regulation by retinoic acid and calcium of transglutaminases in cultured neoplastic and normal human keratinocytes. Cancer Res 46: 2356-2361.
- Rhee JJ, Khuri FF, Shin DM (2004) Advances in chemoprevention of head and neck cancer. Oncologist 9: 302-311.
- Hong WK, Endicott J, Itri LM, Doos W, Batsakis JG, et al. (1986) 13-cis-retinoic acid in the treatment of oral leukoplakia. N Engl J Med 315: 1501-1505.
- Stich HF, Hornby AP, Mathew B, Sankaranarayanan R, Nair MK (1988) Response of oral leukoplakias to the administration of vitamin A. Cancer Lett 40: 93-101.
- Schantz SS (1993) Chemoprevention strategies: the relevance of premalignant and malignant lesions of the upper aerodigestive tract. J Cell Biochem 17: 18-26.
- Benner SE, Pajak TF, Lippman SM, Earley C, Hong WK (1994) Prevention of second primary tumors with isotretinoin in patients with squamous cell carcinoma of the head and neck: long-term follow-up. J Natl Cancer Inst 86: 140-141.
- Figueiredo DL, Mamede RC, Proto-Siqueira R, Neder L, Silva WA Jr, et al. (2006) Expression of cancer testis antigens in head and neck squamous cell carcinomas. Head Neck 28: 614-619.
- Li RJ, Ying X, Zhang Y, Ju RJ, Wang XX, et al. (2011) All-trans retinoic acid stealth liposomes prevent the relapse of breast cancer arising from the cancer stem cells. J Control Release 149: 281-291.
- Lotan R, Xu XC, Lippman SM, Ro JY, Lee JS, et al. (1995) Suppression of retinoic acid receptor-beta in premalignant oral lesions and its up-regulation by isotretinoin. N Engl J Med 332: 1405-1410.
- Gudas LL, Wagner JA (2011) Retinoids regulate stem cell differentiation. J Cell Physiol 226: 322-330.
- Szczepanski MJ, Deleo AB, Luczak M, Molinska-Glura M, Misiak J, et al. (2013) PRAME expression in head and neck cancer correlates with markers of poor prognosis and might help in selecting candidates for retinoid chemoprevention in pre-malignant lesions. Oral Oncol 49: 144-1451.
- Tanaka N, Wang YH, Shiseki M, Takanashi M, Motoji T (2011) Inhibition of PRAME expression causes cell cycle arrest and apoptosis in leukemic cells. Leuk Res 35: 1219-1225.
- Bioley G, Guillaume P, Luescher I, Bhardwaj N, Mears G, et al. (2009) Vaccination with a recombinant protein encoding the tumor-specific antigen NY-ESO-1 elicits an A2/157–165-specific CTL repertoire structurally distinct and of reduced tumor reactivity than that elicited by spontaneous immune responses to NY-ESO-1-expressing tumors. J Immunother 32: 161-168.
- Visus C, Wang Y, Lozano-Leon A, Ferris RL, Silver S, et al. (2011) Targeting ALDH (bright) human carcinoma-initiating cells with ALDH1A1-specific CD8+ T cells. Clin Cancer Res 17: 6174-1684.