Prevention of coronary artery disease (CAD) following a cardiac event has been successful by modifying known conventional risk factors, such as cholesterol. These risk factors are age-dependent and less effective for primary prevention. To halt the pandemic spread of CAD, the number one killer in the world requires primary prevention of cardiac events. 50 percent of all Americans will experience a cardiac event; the challenge is identifying those asymptomatic individuals at risk. Genetics account for 40-60 percent of predisposition for CAD. Since the discovery of the first genetic risk variant, 9p21, in 2007, hundreds of genetic risk variants had been discovered. The genetic burden for CAD can be expressed in a single number, referred to as the Genetic Risk Score (GRS). Retrospective genotyping of clinical trials performed to assess the effect of statin therapy confirmed that GRS improves risk stratification of CAD and detects those benefitting most from statin therapy. In a prospective study of 55,685 individuals, the group with the high GRS had a 91 percent higher risk for cardiac events. Individuals with a healthy lifestyle, and a high GRS, had a 46 percent lower risk for cardiac events in comparison to those with an unfavorable lifestyle. In another trial, risk stratification by GRS showed those at highest genetic risk and performed the most physical activity, had a 50 percent reduction in cardiac events. Genetic risk stratification for CAD has been shown to be superior to conventional risk factors, and the risk is markedly attenuated by lifestyle changes and drug therapy. Genetic risk for CAD can be determined at any time; from birth on since one’s DNA does not change in one’s lifetime. Utilizing the GRS to risk stratify young, asymptomatic individuals could provide a paradigm shift in the primary prevention of CAD and significantly halt its pandemic spread.
coronary artery disease, genetic risk stratification, genetics, primary prevention, genetic risk variants, genomics
Coronary artery disease has reached pandemic proportions and is the number one cause of death throughout the world [1]. In the United States alone, it accounts for over one-third of all deaths [1]. Coronary artery disease refers primarily to atherosclerosis that affects the coronary arteries. This process evolves slowly, which would appear to start very early in life, at least in males. American soldiers, who died during the Vietnamese War, were shown to have fatty streaks in their coronaries, even though the average age was 22 years [2]. These fatty streaks are believed to be the early findings of coronary atherosclerosis, although it is not known for certain whether fatty streaks are reversible or irreversible. It is, however, well recognized that this process increases over the subsequent decades with maximal incidence occurring in males in the late 50s. This increasing trend tends to parallel the increased plasma concentration of low-density lipoprotein cholesterol (LDL-C), which has been shown to be a major culprit in the development of CAD [1,2]. This is illustrated in table 1. The plasma LDL-C in the neonate is about 21-39 mg/dL, which increases to 90 mg/dL in the second decade of life, and to 130-150 mg/dL in the third decade of life [3-5] [Figure 1]. The mean plasma LDL-C in males in the U.S. in their 30s is 146 mg/dL, and for females is 130 mg/dL [6]. Coronary atherosclerosis, while its evolution is gradual, it progresses to coronary endothelial dysfunction, mechanical obstruction, and decreased coronary flow. This process ultimately leads to the clinical manifestations of ischemic heart disease, namely, myocardial infarction, angina, and sudden death. Myocardial infarction is usually preceded by the superimposition of a thrombus onto atherosclerosis, leading to complete coronary occlusion and loss of blood flow to that portion of the myocardium.
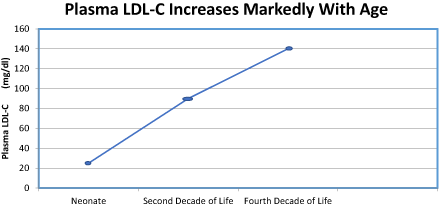
Figure 1. The Marked Increased of Plasma LDL-C with Age
Table 1. Lifetime Changes of LDL-C
Changes of Plasma LDL-C Within a Lifetime |
Neonate |
21-39 mg/dL |
Second Decade |
90 mg/dL |
Third Decade |
130-150 mg/dL |
The evolution of CAD and its sequelae (e.g. myocardial infarction) in females follows a different pattern due to the effect of various hormones in the premenopausal female. Females appear to be relatively protected from CAD until they reach menopause. While the peak incidence of myocardial infarction in males occurs in the late 50s, the peak time of occurrence in the females are in the late 60s [7]. The mechanism remains unknown, whereby these hormones are protective in the premenopausal phase. Replacement of hormones, such as estrogen and progesterone, in the postmenopausal female, does not prevent the development of CAD. Despite this protected pattern in the premenopausal interval, the plasma LDL-C increases to 129 mg/dL in the 30s, as opposed to 146 mg/dL in males [6]. However, once menopause begins, there is a rapid increase in LDL-C, as well as a rapid increase in coronary atherosclerosis. This process is such that within 10 years, the incidence of CAD and cardiac events in women are just as common as it is in males in their 50s [7]. CAD in females, as with males, is the most common cause of death throughout the world. CAD as a cause of death in females is almost 4 times more common than cancer of the breast [8].
Earlier onset of CAD in males or the delay in the onset of CAD in females, and its sequelae, have important implications in the prevention, diagnosis, and treatment of this disease. It appears evident that primary prevention of CAD in the male should begin earlier, probably in the second or third decade of life, while in females perhaps it can be delayed until the fourth or fifth decade of life. To halt this pandemic of CAD, whether it be in males or females, will require primary prevention, as opposed to only secondary prevention. The recent development of the architecture of genetic predisposition to CAD is very exciting; with the potential to detect those at greatest risk prior to the development of CAD. The theme of this review is to outline the role of prediction of CAD based on genetic risk factors and how it may initiate a paradigm shift in the primary prevention of this disease.
Analysis of families and epidemiological studies have for some time claimed that 40-60 percent of predisposition for CAD is inherited [9-11]. The Framingham studies indicated that a family history of CAD was associated with more than a twofold increased risk of CAD in men and women [12]. The prevalence of CAD in monozygotic twins was 44 percent, compared to only 14 percent in dizygotic twins based on the Danish twin registry study [13]. In a recent clinical trial, the INTERHEART study showed a family history of CAD increases the risk for CAD by 1.5 fold [14]. Several recent studies have confirmed that family history is an independent risk factor for CAD [15,16].
The genetic information transmitted from generation to generation is contained in the chromosomes, which are essentially long molecules of DNA. The DNA is a very monotonous molecule made of repeated units of four bases, Adenine, Thymine, Guanine, and Cytosine. It is the sequence of these four bases which confers the unique properties to each individual. The DNA molecule is a double-helix, formed from two complementary strands, with each strand containing 3.2 billion nucleotides, for a total of 6.4 billion nucleotides. Other than identical twins, each human being has unique features attributable to their DNA. Despite the unique variation of each individual genome, the DNA sequence of all humans (homo-sapiens) on this planet is 99 percent identical [17-20]. The corollary to this reflects that only one percent of human DNA is responsible for the unique features comprising each individual. Future endeavors to gain insight into genetic features and genetic predisposition to disease must be targeted to the unique one percent of DNA variation. While one percent of 3.2 billion is 32,000,000, this number is further reduced since most of the 32,000,000 are due to large chunks of DNA that are rearranged, duplicated, or translocated [21]. The majority of DNA variation that determines unique features, such as the color of your hair, the color of your eyes, or predisposition to disease, is due to single nucleotides, referred to as single nucleotide polymorphisms (SNPs) [22]. The number of SNPs present per genome is relatively constant, at about five million. However, it is not that simple since one’s parents select the five million SNPs from billions of SNPs circulating in the general population. Each individual of each generation carries on the average, between 40 and 60 new mutations [23,24], almost all of which are single nucleotides. Given a population of 8 billion, this provides over 400 billion new mutations per generation.
Based on epidemiological studies and current knowledge of DNA, it was expected that genetic predisposition for a disease such as CAD would be carried by multiple genes, each imparting only minimal increased risk. These studies lead to the hypothesis that genetic risk for common chronic diseases, such as CAD, would be due to genetic variants that occur commonly [25]. An unbiased approach to identify genetic risk variants for CAD, or any common disease, was delayed until the development of appropriate technology in about 2005 [26]. In the decade prior to this, there were many claims of genetic risk variants based on the candidate gene approach. Essentially, all of these claims were proven to be incorrect with the development of more appropriate unbiased approaches [27,28]. In the candidate gene approach, one selected a particular gene because of its function and then determined its frequency in a particular disease. This, of course, was a biased selection, and there were no attempts made to replicate the frequency of that gene in an independent population. In 2005, several things evolved to make this possible. An unbiased approach would be to have millions of DNA markers that would span the human genome [25]. One could then compare the frequency of these markers in cases with the disease of concern to that of controls. This is referred to as, case-control association studies [25,29]. Any marker occurring more frequently in the cases than controls would be interpreted to be a genetic risk variant for the disease. The annotation of over a million SNPs by HapMap [30] made it possible to span the human genome with these SNPs as markers to provide an unbiased approach, referred to today as a Genome-Wide Association Study (GWAS) [25]. However, if one is trolling with a million markers, and accept a P-value of 0.05, this would result in 50,000 false positives. This has led to a statistical correction referred to as the Bonferroni, which requires a P-value of 0.000000005 [31]. Furthermore, it is also required that the SNP detecting the genetic risk variant be replicated in an independent population. In addition to the HapMap making available SNPs as markers, platforms were developed to rapidly genotype multiple markers spanning the human genome [26]. This led to algorithms that would rapidly genotype and analyze the frequency of SNPs associated with the disease. We will focus in this review on the results of GWAS in which the cases have documented CAD and the controls have been confirmed on a clinical basis to not have significant CAD [32].
We and the deCode group in Iceland identified simultaneously and independently the first genetic risk variant, 9p21, for CAD in 2007 [33,34]. Shortly thereafter, 9p21 was confirmed by the Wellcome Trust Group [35], and subsequently, multiple investigators confirmed 9p21 as a CAD risk factor in Caucasians [35-37]. 9p21 was shown to be a risk factor for CAD in the Chinese population [38,39], the Korean population [40,41], the Italian population [42], Japanese [41], and also in Southeast Asians [43]. In a more recent study [44] of 30,482 which included 6996 cases of South Asian ancestry from Pakistan and India, 9p21 was confirmed to be a risk factor with a frequency and odds ratio similar to that of Caucasian populations. However, African Americans did not exhibit 9p21 as a risk factor [43].In our pursuit of 9p21we genotyped and phenotyped a sample size of over 23,0000, which included populations from Ottawa, Canada, Houston and Dallas, Texas, and Copenhagen, Denmark [33]. Our results for 9p21 were similar to those of the deCODE group in Iceland [34]. The genetic risk variant was located on the short arm (p) of chromosome 9, which is now commonly referred to as 9p21. The 9p21 risk variant for CAD was associated with a 25 percent increased relative risk per copy and was found to be extremely common, occurring in about 75 percent of the world’s population. It is of note that the risk transmitted by 9p21 is independent of known conventional risk factors such as cholesterol or diabetes. It confirmed the hypothesis that genetic risk variants predisposing to CAD occur commonly. It also emphasized the minimal risk imparted by a single genetic risk variant. The small effect size of the 9p21 variant enhanced the need to have even larger sample sizes. An international consortium was formed among investigators pursuing GWAS for CAD from different countries including the US, Canada, UK, Iceland and Germany [45]. This consortium was referred to as Coronary ARtery DIsease Genome-wide Replication and Meta-analysis (CARDIoGRAM) [45]. Other investigators would also join, leading to a name change to CARDIoGRAMPlusC4D [44]. Over the subsequent decade, this international consortium, the largest collaboration ever in cardiology, together with independent investigators, have discovered hundreds of genetic variants predisposing to CAD [46,47]. The sample size initially was 88,000 cases and controls which rapidly increased to over 200,000.
The genetic risk variants predisposing to CAD provided insight into the genetic architecture of CAD: (1) these genetic risk variants occur extremely commonly, most being present in more than 50 percent of the population. (2) The increased risk was minimal, on average each variant contributed less than 10 percent increased relative risk for CAD. (3) Over two-thirds of the risk variants mediate the risk for CAD independent of known risk factors (cholesterol, diabetes, hypertension, family history, sedimentary lifestyle, age, and smoking). (4) Over 80 percent of the genetic risk variants are in DNA regions that do not code for proteins [Table 2]. The corollary is that these genetic variants manifest their risk for CAD through a regulatory influence on protein-coding genes located upstream or downstream (cis-acting) of the sequence, or even on other chromosomes (trans-acting).
Table 2. The Features of the Genetic Risk Variants for CAD
CAD Genetic Risk Variant Features |
CAD Genetic Risk Variants Occur Extremely Frequently |
Each Genetic Risk Variants Transmits Minimal Risk |
Two-thirds of Genetic Risk Variants for CAD are Independent of Traditional Risk Factors |
Three-fourths of Genetic Risk Variants for CAD Occurs in Regions that do not Code for Proteins |
Since the 1960s, there have been a number of risk factors identified that predisposes an individual to CAD. The most predominant pathogenic factor of coronary atherosclerosis is increased levels of plasma LDL-C. There have been several trials conducted over the last 30 years which showed decreasing plasma LDL-C, was consistently associated with a 30 to 40 percent decrease in the frequency of cardiac events [14,48]. Other risk factors for CAD include diabetes, smoking, age, hypertension, family history, and a sedentary lifestyle. Reduction in all of these factors has consistently shown a significant reduction in cardiac events [48].
It is self-evident from epidemiological studies that comprehensive prevention of CAD would require reducing both acquired and genetic risk factors. The discovery of genetic risk variants predisposing to CAD will provide insight into many aspects of CAD such as pathogenesis but could also serve as biomarkers to predict the risk of this disease. Genetic markers have a major advantage over conventional risk factors in predicting disease since the DNA does not change in one’s lifetime, and thus the risk from genetics is the same at birth as at death. Conventional risk factors such as cholesterol or blood pressure are very age-dependent and become much more reliable as one age. This is illustrated for plasma LDL-C, as shown in table 1. The plasma LDL-C concentration significantly increases with age, as does blood pressure and age itself as a risk factor. Since genetic risk factors are independent of age, they offer a much-improved marker for primary prevention. A major issue in the treatment of a common disease such as CAD is identifying those who are at higher risk and would benefit most from preventive measures. The plasma LDL-C, as stated previously, averages about 121 mg/dL in a female in her 40s, and at 147 mg/dL in males. One may say, why not treat everyone with lifestyle changes and statin therapy early in life? This, of course, must be tempered with the fact that only 50 percent of males or females will develop a cardiac event in their lifetime. It would be most appropriate if one could identify that 25 to 50 percent who would most benefit from preventive therapy. Conventional risk factors are terrific for secondary prevention after someone had a cardiac event but are less than ideal for early primary prevention. The benefit of genetic risk factors is their independence of age and the potential to predict at a very early age those who are at greatest risk of developing CAD.
Each genetic risk variant for CAD contributes only minimally to the increased risk. Based on the more than 300 genetic risk variants for CAD, the average increase in relative risk per variant is less than 10 percent [49]. Thus, the overall genetic risk burden of CAD is proportional to the number of risk variants inherited rather than any single variant. The total genetic risk burden of CAD can be summarized in a single number. The precise odds ratio for each genetic risk variant for CAD has been determined in the genome-wide association studies. Utilizing blood, saliva, or tissue, one can genotype the DNA for the number of genetic variants inherited by each individual. The number of copies for each single genetic risk variant can only vary from 0 to 2. It is 0 if neither of the parents has transmitted the genetic variant, 1 if only one of the parents transmit the genetic variant and 2 if both parents transmit the genetic variant. The risk is accounted for by utilizing the odds ratio determined for each variant in the GWAS. The most common approach to determine the weighted risk is to multiple the numbers of copies of each genetic risk variant times the natural log of the odds ratio [50]. The number resulting from the summation of all of these products is the numerical Genetic Risk Score (GRS).
The initial assessment of the GRS as a means to stratify the risk for CAD took advantage of several clinical trials that had been performed to assess the effect of decreasing plasma LDL-C by statin therapy on cardiac events versus a placebo group. The pioneering study was performed by Mega, et al. in 2015 [51]. The study genotyped a sample size of 48,421 individuals by utilizing 27 genetic risk variants for CAD. The population for the study was comprised of four clinical trials, two of which involved primary prevention, while the other two evaluated secondary prevention. The GRS categorized the population into three different risk groups, low, intermediate, and high. The individuals with the highest GRS, ranking them in the high-risk group, had the most benefit from statin therapy. The GRS was also equally effective at stratifying for the risk of CAD in individuals for either primary or secondary prevention. Furthermore, those with a high GRS required treatment of only 25 individuals with a statin to prevent a single cardiac event. This is in contrast to stratification based on known conventional risk factors, which would require treatment of over 100 individuals. The West of Scotland Coronary Prevention Study (WOSCOPS) was also genotyped and similar results were observed in this study [52]. Those identified as high genetic risk based on GRS displayed a relative risk reduction of 44 percent in comparison to a relative risk reduction of 24 percent in others [52]. In order to prevent a single coronary event with statin medications, the high genetic risk group required treatment of 13 individuals, while the low-risk group required treatment of 38 individuals. Based on the results, GRS was confirmed to have significantly increased discriminatory power for risk stratification of CAD over that of conventional risk factors. Thus, GRS was more effective in identifying individuals who would benefit the most from statin therapy.
Since the initial development of the first genetic risk variant, 9p21, for CAD in 2007 [9,33,34], the pursuit for additional genetic risk variants have been intense. A total of 163 genetic risk variants of genome-wide significance, with replication in an independent population, have been discovered [49]. There are hundreds more that do not reach genome-wide significance but are statistically significant with less than a 5 percent false discovery rate. Nevertheless, if one combines both groups of genetic risk variants, it would only account for 38 percent of inheritability [49], while the estimated inheritability for CAD is in the range of 40-60 percent [9,11]. In an attempt to increase the number of genetic risk variants for CAD, two different approaches were taken. One technique by Inouye, et al. [53] was to include less stringent statistics, such as a false discovery rate of only 5 percent, which resulted in a microarray containing 1.7 million genetic risk variants. Khera, et al. [54] utilized a computerized algorithm referred to as LDpred which predicts genetic variants that associate with a predisposition for CAD. Further pruning was performed to ensure exclusion of linkage disequilibrium, resulting in a microarray with 6.6 million genetic risk variants predisposing to CAD.
The genetic risk score with the markedly increased number of genetic variants, referred to as a polygenic risk score, was assessed in a population provided by the UK biobank. Utilizing a sample size of 288,978 and the 6.6 million microarrays, the analysis showed 8 percent of the population inherited a threefold increased risk for CAD, and 0.5 percent inherited a fivefold risk for CAD. This high-risk group would not have been identified by conventional risk factors for CAD since only 20 percent of the individuals with the threefold increase risk for CAD had hypercholesterolemia and only 28 percent had hypertension. A family history of CAD was present in only 35 percent of the high-risk individuals.
The 1.7 microarrays by Inouye, et al. [53] utilized a sample size of nearly 500,000. The top 20 percent risk group had a fourfold increased risk for CAD. These results from both groups confirmed increased predictive power over that of previous microarrays utilizing less genetic risk variants for CAD.
It is not uncommon in both public and health-related individuals to hold the belief that the events caused by your genes cannot be reversed. This, of course, has been proven to be a myth regarding many diseases, including CAD. It is perhaps worth noting that genes per se, are not the plebiscites that induce changes. Genes provide the blueprint for proteins, which are distributed throughout the cell to perform its various functions. Statins, as an example, inhibit the activity of the rate-limiting enzyme 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) for the synthesis of cholesterol. The gene encoding for this enzyme is not affected by statin therapy, but its effect is inhibited through inhibiting the protein it encodes, HMG-CoA. Nevertheless, it remains to be determined whether individuals stratified to be at high genetic risk for CAD would exhibit a reduction by preventive measures such as lifestyle changes or statin therapy. Cholesterol is the main culprit in the development of coronary atherosclerosis and statin therapy is by far the predominant means of both primary and secondary prevention of CAD. We previously summarized the data from clinical trials that utilized statin therapy, as primary and secondary prevention.
These trials were retrospectively genotyped for genetic risk variants predisposing to CAD and the resulting GRS utilized to risk stratify individuals into high, intermediate, and low risk for CAD. In these trials, individuals ranked at high risk for CAD were also receiving the most statin therapy and exhibited the greatest decrease in cardiac events. Thus, genetic risk stratification of CAD enabled one to identify those at the highest risk and would benefit most from preventive measures, in this case, statin therapy.
Lifestyle changes are always key to the prevention of CAD and are often the first in a series of steps for primary prevention. The first comprehensive clinical trial assessing whether changes in lifestyle can modify genetic risk for CAD was performed recently by Khera, et al. [55]. The sample size consisted of 55,685 participants and the microchip contained 50 genetic risk variants. The endpoint was a favorable lifestyle versus an unfavorable lifestyle. A favorable lifestyle consisted of no current smoking, no obesity, a healthy diet, and frequent exercise, versus a lifestyle that had at least two of these unfavorable components. Risk stratification for CAD was performed following genotyping and development of a genetic risk score. Those in the top 20 percent with a high GRS had a 91 percent higher risk of cardiac events than those with a low GRS. Individuals with a favorable lifestyle, and a high GRS, had a 40 percent lower risk for cardiac events than an unfavorable lifestyle. In a recent study by Tikkanen, et al. [56] genetic risk stratification was used to assess the effect of physical activity on the genetic risk for CAD. The sample size obtained from the UK biobank was 468,095 individuals. The physical activity consisted of a handgrip for 3 seconds and cardiorespiratory fitness determined by oxygen-consumption during cycle ergometer on a stationary bike. The higher level of physical activity was associated with less CAD in each of the low, intermediate, and high genetic risk categories. The highest GRS had the most benefit from cardiorespiratory exercise, with a 49 percent lower risk for CAD [54-56].
Public health authorities throughout the world are recognizing the need for primary prevention to halt the epidemic spread of CAD. Clinical trials have documented repeatedly that reduction in conventional risk factors, such as cholesterol and hypertension, are consistently associated with a reduction in cardiac events. Comprehensive prevention is only possible if we combine genetic and acquired risk factors both for risk stratification and prevention. There is an emerging thrust to develop biobanks so that both the genotype and phenotype can be determined and will provide further insights into this new comprehensive prevention program. Several national efforts are ongoing, which include the UK Biobank, Million Veteran Program, the China Kadoorie Biobank, and the All of Us Research Program in the US. All of these national repositories are aiming for comprehensive data, including genome sequencing of 500,000 to 1 million individuals selected from different ethnic and environmental groups within that country. There are also smaller cohorts being formed, such as the longstanding deCODE Genetics (Iceland), the Danish National Biobank, FinnGen (Finland), Vanderbilt University, Geisinger Health Systems, and Kaiser Permanente Research Bank. These biorepositories will be invaluable in developing wellness and primary prevention programs for a variety of diseases.
We have long known that several risk factors contribute to the predisposition of CAD. These conventional risk factors are primarily acquired or due to environmental exposure. Reducing these risk factors has been very effective in reducing the risk of CAD, along with a reduction in cardiac events. We are now in the process of defining genetic risk factors that are claimed to account for up to 50 percent of predisposition for CAD. Utilizing the current genetic risk variants for CAD, it has been possible to improve risk stratification for CAD and furthermore, have been shown to be reduced by lifestyle changes and drug therapy. The genetic risk was reduced by nearly 50 percent. We are on the eve of comprehensive risk stratification and prevention. Conventional risk factors such as hypocholesteremia, or hypertension, are age-dependent, and become more reliable in the 6th and 7th decade of life, as shown in Table 1. In contrast, genetic risk factors for CAD are independent of age, and thus provide the same risk at birth as anytime thereafter. The risk is innate to the DNA, which does not change in one’s lifetime. The genetic risk score is inexpensive, simple, and can be obtained even from saliva if a blood sample is not available. It can be determined in high, low, and intermediate income countries. Genetic testing, followed by appropriate preventative measures, if induced in females in their 40s, will still be expected to prevent most CAD. Primary prevention in males may be more effective if initiated in the 20s or 30s. It is well recognized from epidemiological studies that around 50 percent of individuals living out a lifespan in the US will experience a cardiac event. The goal for the future is to recognize the 50 percent at risk and initiate primary prevention before the development of CAD and its sequelae. We believe the time is fast approaching, when comprehensive risk stratification for CAD should be inaugurated, followed by appropriate preventative measures. Changes in lifestyle, as well as statin therapy, today are both inexpensive and available throughout the world. Genetic risk stratification for CAD should enable a paradigm shift in the primary prevention of this pandemic disease.
The author has received research funding from the Canadian Institutes of Health Research, CIHR no. MOP P82810, the Canada Foundation for Innovation, the CFI, no. 11966, and the Dignity Health Foundation.
- Murray CJ, Lopez AD (2013) Measuring the global burden of disease. N Engl J Med 369: 448-457. [Crossref]
- Hong YM (2010) Atherosclerotic cardiovascular disease beginning in childhood. Korean Circ J 40: 1-9.
- Kones R, Rumana U (2015) Current treatment of dyslipidemia: a new paradigm for statin drug use and the need for additional therapies. Drugs 75: 1187-1199.
- Fujita H, Okada T, Inami I, Makimoto M, Hosono S, et al. (2008) Low- density lipoprotein profile changes during the neonatal period. J Perinatoly 28: 335-340.
- Pac-Kozuchowska E (2007) Evaluation of lipids, lipoproteins and apolipoproteins concentrations in cord blood serum of newborns from rural and urban environments. Ann Agric Environ Med 14: 25-29.
- Swiger KJ, Martin SS, Blaha MJ (2014) Narrowing sex differences in lipoprotein cholesterol subclasses following mid-life: the very large database of lipids (VLDL-10B). J Am Heart Assoc 3: e000851.
- Nabel EG (2015) Heart disease prevention in young women: Sounding an alarm. Circulation 132: 989-991.
- Canto JG, Kiefe CL (2014) Age-specific analyses of breast cancer versus heart disease mortality in women. Am J Cardio 113: 410-411.
- Zdravkovic S, Wienke A, Pedersen NL, Marenberg ME, Yashin AI, et al. (2002) Heritability of death from coronary heart disease: a 36-year follow-up of 20966 Swedish twins. J Intern Med 252: 247-254.
- Wienke A, Holm NV, Skytthe A, Yashin AI (2001) The heritability of mortality due to heart diseases: a correlated frailty model applied to Danish twins. Twin Res 4: 266-274.
- Marenberg ME, Risch N, Berkman LF (1994) Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med 330: 1141-1146.
- Schildkraut JM, Myers RH, Cupples LA, Kiely DK, Kannel WB (1989) Coronary risk associated with age and sex of parental heart disease in the Framingham Study. Am J Cardiol 64: 555-559. [Crossref]
- Allen G, Harvald B, Shields J (1967) Measures of twin concordance. Acta Genet Stat Med 17: 475-481. [Crossref]
- Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, et al. (2004) Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the interheart study): case-control study. Lancet 364: 937-952.
- Cooper JA, Miller GJ, Humphries SE (2005) A comparison of the PROCAM and Framingham point-scoring systems for estimation of individual risk of coronary heart disease in the Second Northwick Park Heart Study. Atherosclerosis 181: 93-100. [Crossref]
- Nielsen M, Andersson C, Gerds TA, Andersen PK, Jensen TB, et al. (2013) Familial clustering of myocardial infarction in first-degree relatives: a nationwide study. Eur Heart J 34: 1198-1203.
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, et al. (2019) Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun 10: 49-68.
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, et al. (2015) An integrated map of structural variation in 2,504 human genomes. Nature 526: 75-81.
- Chaisson MJ, Wilson RK, Eichler EE (2015) Genetic variation and the de novo assembly of human genomes. Nat Rev Genet 16: 627-640. [Crossref]
- Eichler EE (2019) Genetic variation, comparative genomics, and the diagnosis of disease. N Engl J Med 381: 64-74.
- Roberts R, Campillo A, Schmitt M (2019) Prediction and management of CAD risk based on genetic stratification. Trends Cardiovasc Med 12.S1050-1738: 30115.
- Stranger BE, Forrest MS, Dunning M (2007) Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315: 848-853.
- Sun JX, Helgason A, Masson G (2012) A direct characterization of human mutation based on microsatellites. Nat Genet 44: 1161-1165.
- Kruglyak L, Nickerson DA (2001) Variation is the spice of life. Nat Genet 27: 234-236. [Crossref]
- Hirschhorn JN, Daly MJ (2005) Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6: 95-108. [Crossref]
- Roberts R (2007) New gains in understanding coronary artery disease. Interview with Dr. Robert Roberts. Affymetrix Microarray Bulletin 3: 1-4.
- Morgan TM, Krumholz HM, Lifton RP, Spertus JA (2007) Nonvalidation of reported genetic risk factors for acute coronary syndrome in a large-scale replication study. JAMA 297: 1551-1561.
- Anand (2009) Genetic variants associated with myocardial infarction risk factors in over 8,000 individuals from five ethnic groups: INTERHEART Study. Circ Cardiovas Genet 2: 16-25.
- Roberts R (2008) Personalized Medicine: A Reality Within this Decade. J of Cardiovasc Trans Res 1: 11-16.
- Gibbs RA (2003) The International HapMap Project. Nature 426: 789-779.
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273: 1516-1517
- Roberts R, Stewart AF, Wells GA, Williams KA, Kavaslar N, et al. (2007) Identifying genes for coronary artery disease: An idea whose time has come. Can J Cardiol 23 Suppl A: 7A-15A. [Crossref]
- McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, et al. (2007) A common allele on chromosome 9 associated with coronary heart disease. Science 316: 1488-1491.
- Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, et al. (2007) A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 316: 1491-1493.
- Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661-678.
- Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, Novartis Institutes of BioMedical Research, Saxena R, Voight BF, Lyssenko V, et al. (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316: 1331-1336.
- Broadbent HM, Peden JF, Lorkowski S, Goel A, Ongen H, et al. (2008) Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet 17: 806-814.
- Ding H, Xu Y, Wang X, Wang Q, Zhang L, et al. (2009) 9p21 is a shared susceptibility locus strongly for coronary artery disease and weakly for ischemic stroke in Chinese Han population. Circ Cardiovasc Genet 2: 338-346.
- Zhou L, Zhang X, He M, Cheng L, Chen Y, et al. (2008) Associations between single nucleotide polymorphisms on chromosome 9p21 and risk of coronary heart disease in Chinese Han population. Arterioscler Thromb Vasc Biol 28: 2085-2089.
- Shen GQ, Li L, Rao S, Abdullah KG, Ban JM, Lee BS, et al. Four SNPs on chromosome 9p21 in a South Korean population implicate a genetic locus that confers high cross-race risk for development of coronary artery disease. Arterioscler Thromb Vasc Biol 28: 360-365.
- Hinohara K, Nakajima T, Takahashi M, Hohda S, Sasaoka T, et al. (2008) Replication of the association between a chromosome 9p21 polymorphism and coronary artery disease in Japanese and Korean populations. J Hum Genet 53: 357-359.
- Shen GQ, Rao S, Martinelli N, Li L, Olivieri O, et al. (2007) Association between four SNPs on chromosome 9p21 and myocardial infarction is replicated in an Italian population. J Hum Genet 53: 144-150.
- Assimes TL, Knowles JW, Basu A, Iribarren C, Southwick A, et al. (2008) Susceptibility locus for clinical and subclinical coronary artery disease at chromosome 9p21 in the multi-ethnic ADVANCE study. Hum Mol Genet 17: 2320-2328.
- The Coronary Artery Disease (C4D) Genetics Consortium (2011) A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet 43: 339-344.
- Preuss M, König IR, Thompson JR (2010) CARDIoGRAM Consortium. Design of the Coronary ARtery DIsease Genome-Wide Replication And Meta-Analysis (CARDIoGRAM) Study: a genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circ Cardiovasc Genet 3: 475-483.
- Assimes TL, Roberts R (2016) Genetics: implications for prevention and management of coronary artery disease. J Am Coll Cardiol 68: 2797-2818.
- Musunuru K, Kathiresan S (2019) Genetics of Common, Complex Coronary Artery Disease. Cell 177: 132-145. [Crossref]
- Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, et al. (1998) Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 279: 1615-1622. [Crossref]
- Erdmann J, Kessler T, Munoz Venegas L, Schunkert H (2018) A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res 11: 1241-1257.
- Goldstein BA, Knowles JW, Salfati E, Ioannidis JP, Assimes TL (2014) Simple, standardized incorporation of genetic risk into non-genetic risk prediction tools for complex traits: coronary heart disease as an example. Front Genet 5: 254.
- Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield M, et al. (2015) Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 385: 2264-2271.
- Natarajan P, Young R, Stitziel NO, Padmanabhan S, Baber U, et al. (2017) Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 135: 2091-2101.
- Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, et al. (2018) Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J Am Coll Cardiol 72: 1883-1893. [Crossref]
- Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50: 1219-1224.
- Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, et al. (2016) Genetic Risk, Adherence to a Healthy Lifestyle, and Coronary Disease. N Engl J Med 375: 2349-2358. [Crossref]
- Tikkanen E, Gustafsson S, Ingelsson E (2018) Associations of fitness, physical activity, strength, and genetic risk with cardiovascular disease: longitudinal analyses in the UK Biobank study. Circulation 137: 2583-2591.