Protein Z-dependent protease inhibitor (ZPI), a serpin family protein, together with its tightly bound vitamin K-dependent protein cofactor, protein Z (PZ), function to rapidly and specifically inhibit membrane-bound FXa. Along with antithrombin, tissue factor pathway inhibitor (TFPI), and activated protein C (APC), the ZPI/PZ complex comprises one of the major endogenous anticoagulants in human plasma. Recent findings showing that ZPI/PZ regulates prothrombinase activity and that ZPI/PZ gene knockout partly restores hemostasis in a hemophilia mouse model have suggested that the neutralization of ZPI/PZ anticoagulant function could provide an alternative therapy for hemophilia treatment. Compared with other alternative therapies currently in development that target APC, TFPI, and antithrombin anticoagulants, anti-ZPI/PZ therapy is expected to be safer and similarly effective. Recent progress in the development of potent, specific ZPI/PZ antagonists should enable testing of this potentially safer therapy in a hemophilia mouse model.
protein z-dependent protease inhibitor, protein z, hemophilia, serpin, endogenous anticoagulant, blood coagulation
Protein Z-dependent protease inhibitor (ZPI) was first identified and purified from plasma as a Protein Z (PZ)-dependent inhibitor of active factor X (FXa) [1], and later identified as a member of the serpin superfamily (serpinA10) [2]. ZPI circulates in plasma as a tight complex with the vitamin K-dependent protein Z (PZ) (Kd ~10 nM), which is structurally homologous to factors II, VII, IX, X, but lacking protease activity due to the absence of serine and histidine residues of the catalytic triad [3]. In human plasma, ZPI and PZ levels cover a broad range (ZPI ~59±8 nM, PZ ~46±16 nM) [4,5], with ZPI in modest excess over PZ; thus most ZPI and PZ exist as the complex in plasma [6]. The plasma ZPI/PZ level was found to be increased by oral contraceptive use and decreased by oral anticoagulant therapy [5,7]. Plasma deficiencies in ZPI or PZ were found to be a risk factor for vascular thrombosis by several studies but an uncertain risk factor by others [8].
ZPI/PZ comprises one of four major anticoagulant systems in plasma. The other three include: antithrombin which inhibits FXa, active factor IX (FIXa), and thrombin; activated protein C (APC) which proteolytically degrades active factor V (FVa) and active factor VIII (FVIIIa); and tissue factor pathway inhibitor (TFPI) which inhibits the extrinsic FXase complex (TF-VIIa-FXa) and FXa. ZPI/PZ functions as an anticoagulant by specifically inhibiting membrane associated FXa. ZPI also inhibits active factor XI (FXIa) but this inhibition does not require PZ [9], and its importance for ZPI anticoagulant function remains unclear.
The importance of ZPI/PZ in the regulation of blood coagulation is indicated from findings that ZPI or PZ deficiencies in patients or mouse models when combined with the factor V Leiden (FVL) mutation result in a severe or fatal thrombosis phenotype [10-13]. More recent studies have suggested that such prothrombotic states arise from impairments in the regulation of FXa in prothrombinase by ZPI/PZ and the complementary regulation of FVa in prothrombinase by APC [14] (Figure 1).
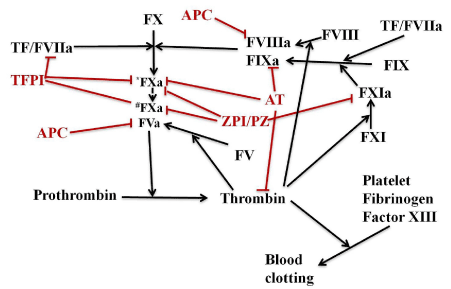
Figure 1. Schematic of major anticoagulant systems in plasma that regulate thrombin generation. Blood coagulation is initiated by the extrinsic Xase complex (TF-VIIa), which rapidly activates FX to FXa and FIX to FIXa. FIXa binds FVIIIa to form the intrinsic Xase complex which amplifies FX activation. FXa binds FVa to form prothrombinase (FXa-FVa) which activates prothrombin to thrombin. Deficiencies in the intrinsic Xase complex (FIXa or FVIIIa) result in hemophilia. The major anticoagulant regulators of thrombin formation include: ZPI/PZ which inhibits free FXa (*FXa) and prothrombinase bound FXa(#FXa); antithrombin (AT) which inhibits thrombin, FIXa and free FXa; APC which proteolytically degrades FVa and FVIIIa; and TFPI which inhibits the extrinsic Xase complex and prothrombinase bound FXa. Alternative strategies to FVIII/FIX replacement for treating hemophilias involve targeting APC, antithrombin, TFPI and ZPI/PZ anticoagulants
The therapies targeting anticoagulant systems for hemophilia treatment
Alternative approaches to treat hemophilia have focused on down-regulating anticoagulant systems including TFPI, antithrombin or APC. Such approaches are currently at different stages of development. Two humanized anti-TFPI monoclonal antibodies are undergoing clinical testing: concizumab from Novo Nordisk is being evaluated in a phase 2 study [15] and Bay 1093884 from Bayer is being tested in an initial human dosage trial [16]. ALN-AT3 (fitusiran from Alnylam Pharmaceuticals), an RNAi that targets antithrombin, is currently in a phase 1 trial [17]. An APC inhibitor engineered from the serpin, α1-antitrypsin (α1AT), has been found to ameliorate hemophilia mouse bleeding and is still at a laboratory investigation stage [18]. The ZPI/PZ anticoagulant system has been recently proposed and proven to be a similarly effective target for hemophilia treatment. Importantly, the more essential roles that TFPI, APC, and AT play in regulating blood regulation suggest that targeting ZPI/PZ for hemophilia treatment may be expected to be advantageous in many aspects, especially with the concern of safety. This review summarizes the recent progress of understanding the mechanism of ZPI/PZ anticoagulant function and how targeting ZPI/PZ could provide a promising safe prophylactic therapy for hemophilia.
ZPI/PZ Inhibits both free and prothrombinase-bound FXa
Without cofactors, ZPI inhibits FXa with a slow rate constant ~104M-1s-1. However, in the presence of PZ, phospholipids, and Ca2+, the inhibition is accelerated ~1000-fold (~107M-1s-1), close to the diffusion limit. Previous studies have established that the ZPI/PZ complex inhibits FXa bound to a procoagulant phospholipid membrane surface by binding to the membrane through the PZ gamma-carboxyglutamic (Gla) domain, so as to present the ZPI reactive center loop (RCL) to FXa to form a ZPI/PZ-FXa ternary Michaelis complex. The Michaelis complex then undergoes acylation and conformational trapping to form a covalent complex that inactivates the protease’s catalytic function in accord with the standard suicide substrate mechanism of serpin inhibition of coagulation proteases (10). PZ is limiting in plasma and its levels are maintained by functioning catalytically to promote ZPI inhibition of FXa, i.e., by dissociating from ZPI once the ZPI-FXa complex is formed [9]. Unlike other serpin- protease complexes, the ZPI-FXa covalent complex is unstable under the physiological conditions of neutral pH and dissociates with a half-life of ~1 hour to release active FXa and cleaved ZPI [9].
ZPI has a P1 tyrosine (Y387) bait residue in its RCL that is unfavorable for recognition of FXa. As a result, both the RCL and exosites circumscribing the RCL play important roles in the ability of ZPI to specifically recognize FXa. In particular, an interaction of Glu313 in sheet 2C of ZPI with Arg143 of the FXa autolysis loop is essential in mediating the ZPI-FXa interaction [19,20]. Interactions between the Gla domains of PZ and FXa further promote the formation of the ternary ZPI/PZ-FXa Michaelis complex on a procoagulant membrane (Figure 2).
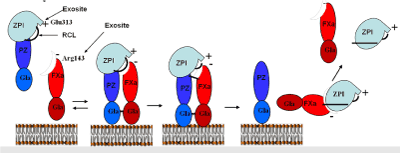
Figure 2. Mechanism of the PZ-dependent inhibition of membrane-bound FXa by ZPI. ZPI/PZ complex binds a phospholipid membrane through the PZ Gla domain and presents the ZPI reactive center loop (RCL) to the active site of membrane-bound FXa to form a ternary Michaelis complex. The Michaelis complex interaction is enhanced by exosite interactions including the Glu313-Arg143 interaction between ZPI and the FXa catalytic domain and Gla-Gla domain interactions between PZ and FXa. The FXa catalytic Ser then attacks the ZPI P1 Tyr to cleave the RCL and form a covalent acyl complex with FXa that is then stabilized by a ZPI conformational change through the standard serpin mechanism. PZ dissociates from the ZPI-FXa covalent complex, which then itself slowly dissociates to form cleaved ZPI and active FXa
Early findings suggested that ZPI/PZ can only inhibit FXa before FV is activated to FVa to form prothrombinase, i.e., FXa in prothrombinase is resistant to ZPI/PZ inhibition [21]. However, recent kinetic studies have shown that prothrombinase- complexed FXa is inhibited by ZPI/PZ at a physiologically significant rate. Although this rate (106M-1s-1) is ~6-8 fold slower than the inhibition of uncomplexed FXa [14], it is still rapid enough to be physiologically relevant given the near diffusion-limited rate of inhibition of uncomplexed FXa on a membrane [14]. Additional kinetic studies of ZPI/PZ inhibition of prothrombinase in the presence of the substrate, prothrombin, showed a modest 3-4-fold further rate reduction, consistent with a dynamic mode of regulation of FXa in prothrombinase that is determined by competing interactions of prothrombin and the ZPI-PZ complex with FXa. The physiologic relevance of ZPI-PZ inhibition of prothrombinase is most evident from the ability of physiologic levels of wild-type (WT) ZPI-PZ complex but not a disabled exosite mutant (Glu313Ala) ZPI-PZ complex to inhibit thrombin generation during prothrombinase-catalyzed activation of physiologic levels of prothrombin (Figure 3). This inhibition becomes evident only when full progress curves of thrombin generation are examined, thereby explaining the failure of the earlier study which followed thrombin generation only in the initial phase to observe a significant inhibitory effect of ZPI/PZ. Together, these data indicate that the ZPI/PZ complex targets FXa both before and after its incorporation into prothrombinase [14]. ZPI/PZ thus appears to complement the APC system in inhibiting prothrombinase during the propagation phase of coagulation [14]. This contrasts with another FXa inhibitor, antithrombin, which is a poor inhibitor of FXa after its incorporation into prothrombinase [22]. Notably, recent studies have shown that TFPI-α also inhibits prothrombinase-bound FXa, but only at an intermediate stage of prothrombinase formation during the initiation phase of blood coagulation [23].
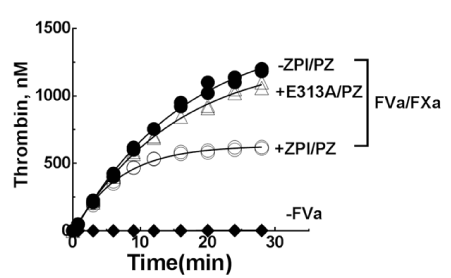
Figure 3. Plasma levels of wild-type or E313A ZPI-PZ complexes inhibit the activation of prothrombin. Time course of prothrombin activation (1.4 µM) by prothrombinase assembled with FVa (16nM), FXa (0.06nM), 25µM 30% PS/70 %PC vesicles, and 5mM Ca2+ in the absence (solid symbols) or presence (open symbols) of 60 nM ZPI or E313A ZPI and 50 nM PZ in I 0.15, pH 7.4 buffer at 25°C. Reactions were initiated with FXa/FVa/lipid/Ca2+ and quenched at varying times with a chromogenic thrombin substrate plus 10 mM EDTA in reaction buffer and residual thrombin activity was determined. Data represent the average of 3–4 independent measurements, mean±S.D. PS: Phosphatidylserine; PC:Phosphatidylcholine. Solid lines are empirical fits of data. Modified from Huang X, et al. [14]
The importance of ZPI/PZ regulation of free and prothrombinase-bound FXa is also indirectly suggested from findings that ZPI and PZ are up-regulated in malignant tumors and colocalized with FX in many cancerous tissues, possibly reflecting a coordinate up-regulation of ZPI/PZ and FX in the tumors [24]. Many coagulation or anticoagulation factors are similarly reported to be up-regulated in malignant tumors [25], and to contribute to a prothrombotic state (see the review by Levi [26]).
ZPI/PZ, factor V Leiden (FVL) and hemophilia
It is known that the FVL mutation, a common FV mutation producing FVa resistant to anticoagulant inactivation by APC, is associated with a moderate prothrombotic risk [27,28] and resembles the ZPI or PZ deficiency phenotype in humans and animal models [11,29]. However, FVL combined with ZPI or PZ deficiency results in a more severe prothrombotic phenotype [10-13]. In contrast, ZPI and or PZ deficiencies have much less influence on other types of prothrombotic risks such as prothrombin G20210A and hyperhomocysteinemia [30]. A plausible mechanism that explains these observations is that APC and ZPI/PZ function as complementary regulators of prothrombinase activity, either by degrading FVa or inhibiting FXa, respectively. Thus, the moderate prothrombotic phenotype observed in FVL or ZPI/PZ deficiency clinically in humans or in animal models suggests that down-regulating one of these anticoagulants causes a modest overactivity of prothrombinase. However, the more severe thrombosis phenotype when FVL is combined with ZPI or PZ deficiency suggests that down- regulating both APC and ZPI/PZ anticoagulants causes a marked overactivity of prothrombinase. Such observations also imply that FVL and ZPI/PZ deficiency are similar in potency to promote a prothrombotic phenotype.
The FVL phenotype is associated with milder clinical symptoms in hemophilia patients including fewer bleeding episodes and a lesser need for coagulation factor transfusions, as reported by many investigators [31-33], (see reviews by van Dijk et al. [34] and Franchini et al. [35], with the molecular mechanism elucidated by Mann and Mertens research groups [36,37]. In addition, in vivo studies with hemophilia mice have clearly shown that the FVL mutation significantly improves hemophilia A and B coagulation test profiles as well as hemostasis following microcirculation injury in both hemophilia A and B mice, comparable to the restoration of hemostasis produced by replenishing FVIII and FIX coagulation factors [38].
Since FV deficiency is lethal, it is difficult to target FV for hemophilia treatment. However, the ZPI/PZ system is a much more feasible target as evidenced by recent developments of ZPI/PZ antagonists [39].
Targeting ZPI/PZ for hemophilia treatment
The anticoagulant regulation of both free and prothrombinase-bound FXa by the ZPI/PZ complex suggests that ZPI/PZ could be a therapeutically important target to regulate hemostasis. Consistent with this hypothesis, a recent study by Thomas Girard and George Broze showed that down-regulation of ZPI or PZ in human hemophilia plasma with an anti-PZ monoclonal antibody or in vivo gene knockout of ZPI or PZ in a hemophilia A mouse model improves the bleeding phenotype significantly, equivalent to
~15% FVIII replacement [40]. It is known that the clinical hemophilia phenotype varies from severe (<1% FVIII), which is associated with spontaneous bleeding, to mild (5-40% FVIII) in which hemorrhage is uncommon in the absence of trauma or surgical procedures [41]. It therefore suggests that neutralizing ZPI/PZ anticoagulant function, though modest in effect, is sufficient to improve the phenotype of severe hemophilia.
Bolkun et al. reported that up-regulation of the ZPI/PZ anticoagulation system was observed in hemophilia A patients (plasma level increases of 19% for ZPI and 26% for PZ) and correlations of ZPI or PZ levels with bleeding rate and joint bleeds were found in severe hemophilia A patients [4].
ZPI/PZ interaction and the development of ZPI/PZ antagonists
The crystal structure of the ZPI/PZ complex has been determined by two groups [20,42] (PDB: 3H5C and 3F1S). These structures, together with mutagenesis studies, have shown that the ZPI-PZ interaction is mediated by ionic interactions of D74, D238, and D293 on helix G and helix A of ZPI with the pseudocatalytic domain of PZ, and hydrophobic interactions of Y240 of ZPI with a cavity formed between the PZ EGF2 and pseudocatalytic domains of PZ (Figure 4). D293 and Y240 account for most of the ZPI- PZ binding energy, and are thus hot spots for ZPI binding to PZ. Importantly, K239, which is one of the ZPI contact residues next to Y240, was found to antagonize the ZPI- PZ interaction. Lys239 to alanine or cysteine mutations resulted in ZPI mutants that bound PZ over 20-fold tighter than WT ZPI. The enhanced binding results either from the smaller neutral sidechains removing a steric blockade or neutralizing a repulsive interaction of the positively charged ZPI K239 sidechain with the nearby basic PZ residues, so as to strengthen the critical hydrophobic interaction between ZPI Y240 and PZ [43,44].
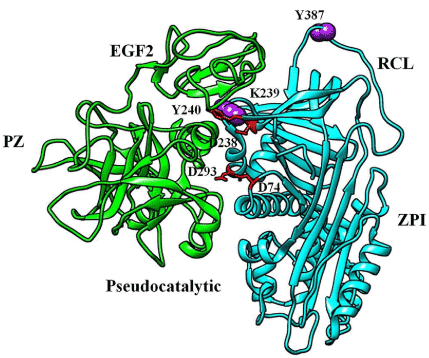
Figure 4. Crystal structure of the ZPI (cyan)-PZ (green) complex (PDB:3H5C). The mutated amino acids are in purple sphere. ZPI residues that bind PZ are in red stick
Based on these findings, a recent study by us engineered two mutations in ZPI, one in the RCL P1 Y387 residue to inactivate the FXa/FXIa inhibitory function of the serpin, and the second in the K239 binding interface residue to enhance the affinity of the inactive ZPI for PZ (Figure 4). The study found that the Y387A/K239A mutant ZPI (ZPI-2A) efficiently competes with and displaces WT ZPI from its complex with PZ to result in inactive ZPI-2A-PZ complexes and uncomplexed WT ZPI. A series of assays were performed to characterize this ZPI mutant which found that [39]: i) ZPI-2A bound PZ >20-fold tighter than WT ZPI (Kd 0.4±0.1 nM vs.10±1 nM); ii) ZPI-2A effectively neutralized ZPI/PZ anti-FXa activity with a ~3-fold molar excess over WT ZPI whether FXa was bound to FVa in prothrombinase or unbound; iii) ZPI/PZ anticoagulant activity was reversed by ZPI-2A in a dose-dependent manner in thrombin generation assays in a purified system or in normal/hemophilia plasmas, with a ~3-fold molar excess of ZPI-2A over WT ZPI being sufficient to fully reverse ZPI/PZ inhibition of thrombin generation (Figure 5). Human ZPI/PZ has a plasma concentration of ~50-60nM, thus full inactivation could be achieved with as low as ~150-200nM ZPI-2A mutant. ZPI-2A thus is a newly developed potent and specific antagonist of ZPI/PZ anticoagulant function. Whether ZPI-2A can ameliorate bleeding in the hemophilia mouse model is now under investigation in the author’s laboratory.
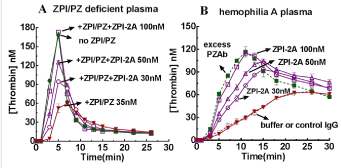
Figure 5. ZPI-2A reverses the anticoagulant effect of ZPI/PZ on thrombin generation in plasma. Reactions were performed in ZPI/PZ-deficient plasma primed with plasma levels of WT ZPI and PZ (A), or in hemophilia A plasma (100μl) in the absence and presence of the indicated final concentrations of ZPI-2A, or anti-PZ polyclonal antibody (PZAb) (0-200μg/ml)/control sheep IgG (B) in I 0.15 Tris buffer pH 7.4 at 37oC. Coagulation was activated by adding small unilamellar lipid vesicles (SUVs) containing 20%PS:20%PE:60%PC, and Ca2+, corn trypsin inhibitor (CTI) (to block contact activation) and tissue factor (TF) to a total volume of 150μl. Final concentrations were 25μM SUVs, 16mM Ca2+, 50μg/ml CTI, 2pM TF, 35nM ZPI/PZ, 0-100nM ZPI-2A, 200µg/ml PZAb/control IgG. All data represent the average of 3-4 independent measurement ± SD. PS: Phosphatidylserine; PE: Phosphatidylethanolamine; PC:Phosphatidylcholine. Modified from Huang X, et al. [39]
Another possible strategy of down-regulating ZPI/PZ activity by an antagonist is to identify a small molecule that can bind to the ZPI/PZ interface and disrupt the ZPI/PZ complex. Recent progress in the discovery and development of small molecule antagonists of protein-protein interactions has been encouraging, particularly in the case of small molecules that target binding hotspots of these interactions. The ZPI-PZ interface appears to be especially well suited for this approach, given that ZPI-Y240 forms a hotspot interaction with a largely hydrophobic pocket in its PZ binding partner (Figure 4). This pocket leads to an extended channel through PZ that could form an extended binding site for an inhibitor anchored in the Y240 binding site. A high- throughput screening assay for such a small molecule antagonist could exploit an established ZPI/PZ binding assay which employs a fluorophore-labeled K239C ZPI mutant that sensitively reports PZ binding by undergoing a 300-400% fluorescence enhancement [43]. A small molecule that binds to the ZPI/PZ interface and blocks the formation of the ZPI/PZ complex would thus be identifiable by the quenching of the enhanced fluorescence signal. Unpublished studies of the author have in fact used this assay to screen large libraries and have successfully identified several small molecule antagonists of the ZPI/PZ interaction, although with low affinity and specificity. The advantages of a small molecule drug for hemophilia include: i) it will not cause broad immune responses from subjects, leading to patients becoming refractory to treatment; ii) a small molecule drug can be administered orally, whereas all current therapies are injection-based, and iii) a small molecule is much easier and less expensive to produce. The potential to reduce bleeding in hemophilia patients with an oral medication could be life changing. While efforts to identify a small molecule that specifically binds to ZPI or PZ with high affinity could be challenging, it is a worthy pursuit given the high payoff.
There are many polyclonal or monoclonal anti-ZPI or anti-PZ antibodies commercially available. An anti-PZ polyclonal antibody was found to effectively block ZPI/PZ anticoagulant activity and enhance thrombin generation in human normal and hemophilia plasma [14,39]. Moreover, a monoclonal antibody targeting the human PZ pseudocatalytic domain was found to neutralize ZPI/PZ activity in a recent study, though with a relatively low affinity [40]. Interestingly, autoimmune anti-PZ antibodies have also been detected in some female patients with obstetrical complications including recurrent miscarriage, stillbirth, preeclampsia, intrauterine growth restriction, and placental abruption [45]. It is believed that some of these anti-PZ antibodies would neutralize the ZPI/PZ activity of patients, thus further enhancing the procoagulant states of the patients who already have preexisting hypercoagulation situations due to pregnancy, contributing to abnormal fetal development or fetal loss. Although pathological in these patients, these fully-humanized PZ antibodies (monoclonal), if successfully cloned, expressed, purified and characterized, would have the potential to be developed into useful anti-ZPI/PZ antagonists for clinical use, just as other monoclonal antibodies isolated from human B cells are being developed into useful antibody therapies [46].
The rationale and potential advantages of anti-ZPI/PZ therapy for hemophilia treatment
Targeting APC, antithrombin, or TFPI for hemophilia treatment requires careful dosing to avoid the risk of thrombosis because of the essential roles that these anticoagulants play in maintaining hemostasis, e,g., concizumab is a monoclonal antibody therapy which requires a daily injection with low doses [15]. The long-term safety of these still developing therapies is also unknown. Compared with targeting TFPI, APC and antithrombin, targeting ZPI/PZ is expected to exhibit a much better safety profile. ZPI/PZ is not involved in anti-inflammation and cytoprotection functions as is the case with APC and antithrombin [47-49]. Moreover, ZPI and/or PZ deficiencies in humans are only mildly prothrombotic or normal [8]. In healthy individuals, PZ varies over a wide range with no trace of abnormality in the low PZ population [5]. The association of ZPI and/or PZ deficiencies with thrombosis in clinical patients is often controversial and usually requires other preexisting hypercoagulation conditions like pregnancy, atherosclerosis, etc. [8,45]. Importantly, animals in which ZPI or PZ genes are knocked out are normal and healthy [11]. By contrast, mouse gene deletion of antithrombin, TFPI, or PC causes death in utero or in the perinatal period from thrombosis, disseminated intravascular coagulation, or a consumptive coagulopathy [50-52]. Concomitant hemophilia does not rescue TFPI-deficient [53] or antithrombin-deficient mice [51,54]. Clinical PC and antithrombin deficiencies in humans result in severe thrombosis [55,56]. Safety and health concerns are thus likely to be more problematic in TFPI, antithrombin, and APC targeted therapies that are still under development.
Because of the modest effect of ZPI/PZ neutralization on hemostasis, ZPI/PZ full inactivation does not result in severe thrombosis. The recently published work has shown that full ZPI/PZ inactivation and a concomitant enhanced thrombin generation can be achieved in human hemophilia plasma with the inactive ZPI-2A mutant at low stoichiometric doses. Since ZPI-2A is a variant of WT ZPI with high potency and specificity for PZ binding, it is expected to be low in immunogenicity and to produce fewer side effects and thus be an ideal agent for clinical use. As a serpin, ZPI has a relatively long half-life in plasma (~3 days) similar to the 4.5 day half-life of its most homologous family member α1-AT (serpin A1) [3,57], considerably longer than the half-lives of recombinant FVIII and FIX used for replacement therapy in hemophilias A and B of ~12 hours and 18-24 hours, respectively [58]. ZPI-2A may also be administered subcutaneously, like other serpin medications, e.g. C1 Esterase Inhibitor (C1-INH, serpinG1) (Haegarda), is an approved subcutaneous injection treatment for hereditary angioedema. Whether the efficacy of ZPI/PZ gene knockout in hemophilia mice can be replicated by the administration of mouse ZPI-2A to the hemophilia mouse will be a critical test of the potential of anti-ZPI/PZ therapy. An advantage of anti-ZPI/PZ therapy is that it should be effective in both hemophilia A and B patients, in contrast to a recently approved antibody mimetic of FVIII (emicizumab) which is only effective in treating hemophilia A. The effectiveness of anti-ZPI/PZ therapy in hemophilia patients could be tested by plasma thrombin generation assays that effectively reflect the bleeding tendency of hemophilia A or B patients. However, anti-ZPI/PZ therapy should be more cautiously used or avoided in hemophilia patients with FVL, although this is a very rare situation.
Although studies of the contribution of ZPI/PZ to hemostasis and bleeding in hemophilia patients are still limited, current in vivo and in vitro data support the idea that inhibition of ZPI/PZ anticoagulant function could potentially be an effective and safer prophylactic therapy for hemophilia patients, similar to the clinical observation that FVL ameliorates the hemophilia bleeding phenotype. Such therapy could partially restore the defective hemostasis system of hemophilia patients, and tremendously improve hemophilia treatment because most hemophilia bleeding is not trauma-induced but results from spontaneous microcirculation injury. This is particularly the case with severe hemophilia that accounts for 60% of the hemophilia population. In addition, when administered as an adjuvant therapy, such therapy could significantly reduce the frequency of coagulation factor replacement, which is problematic for some patients (25-30%), due to inhibitor development arising from stimulation of the immune system [59]. It should be noted that although gene therapy for hemophilia has made great progress in recent years, 30~50% of patients are not eligible for such treatment because of the pre- existence of antibodies against the virus vector used for gene therapy [60]. Due to liver toxicity, patients with liver disease, which is common in hemophilia patients > 35 years old due to pathogen containing plasma transfusions in the past, will also be ineligiblefor gene therapy [60], underscoring the necessity to develop other new therapies. Most encouragingly, recent progress in the development of novel ZPI/PZ antagonists should now enable investigators to test this novel therapy in the hemophilia mouse model.
Funding for this work was provided by National Institutes of Health Grants R37HL39888 (to Drs. Steven Olson and Xin Huang (Co-PI)). I thank Dr. Steven T. Olson of the Department of Periodontics at the University of Illinois at Chicago for helpful discussions of this work and for help with editing the manuscript.
The author declares no conflicts of interest with the contents of this article.
- Han X, Fiehler R, Broze GJ (1998) Jr. Isolation of a protein Z-dependent plasma protease inhibitor. Proceedings of the National Academy of Sciences of the United States of America 95: 9250-9255.
- Han X, Huang ZF, Fiehler R, Broze GJ (1999) The protein Z-dependent protease inhibitor is a serpin. Biochemistry 38: 11073-11078.
- Broze GJ, Miletich JP (1984) Human Protein Z. The Journal of Clinical Investigation 73: 933-938.
- Bolkun L, Galar M, Piszcz J, Lemancewicz D, Kloczko J, et al. (2013) Plasma concentration of protein Z and protein Z-dependent protease inhibitor in patients with haemophilia A. Thrombosis Research 131: e110-e113.
- Miletich JP, Broze GJ (1987) Human plasma protein Z antigen: range in normal subjects and effect of warfarin therapy. Blood 69: 1580-1586.
- Tabatabai A, Fiehler R, Broze GJ (2001) Protein Z circulates in plasma in a complex with protein Z-dependent protease inhibitor. Thrombosis and Haemostasis 85: 655-660.
- Al-Shanqeeti A, van Hylckama Vlieg A, Berntorp E, Rosendaal FR, Broze GJ, et al. (2005) Protein Z and protein Z-dependent protease inhibitor. Determinants of levels and risk of venous thrombosis. Thrombosis and Haemostasis 93: 411-413.
- Corral J, Gonzalez-Conejero R, Hernandez-Espinosa D, Vicente V (2007) Protein Z/Z-dependent protease inhibitor (PZ/ZPI) anticoagulant system and thrombosis. British journal of Haematology 137: 99-108.
- Huang X, Swanson R, Broze GJ, Olson ST (2008) Kinetic characterization of the protein Z-dependent protease inhibitor reaction with blood coagulation factor Xa. The Journal of Biological Chemistry 283: 29770-297783.
- Yin ZF, Huang ZF, Cui J, Fiehler R, Lasky N, et al. (2000) Prothrombotic phenotype of protein Z deficiency. Proceedings of the National Academy of Sciences of the United States of America 97: 6734-6738.
- Zhang J, Tu Y, Lu L, Lasky N, Broze GJ, et al. (2008) Protein Z-dependent protease inhibitor deficiency produces a more severe murine phenotype than protein Z deficiency. Blood 111: 4973-4978.
- Kemkes-Matthes B, Nees M, Kuhnel G, Matzdorff A, Matthes KJ, et al. (2002) Protein Z influences the prothrombotic phenotype in Factor V Leiden patients. Thrombosis Research 106: 183-185.
- Kemkes-Matthes B MK, Souri M, Koseki-Kuno S, Ichinose A (2005) R255h amino acid substitution of protein Z identified in patients with factor V Leiden mutation. Br J Haematol 128: 248-252.
- Huang X, Swanson R, Kroh HK, Bock PE (2019) Protein Z-dependent protease inhibitor (ZPI) is a physiologically significant inhibitor of prothrombinase function. The Journal of Biological Chemistry 294: 7644-7657.
- Shapiro AD, Angchaisuksiri P, Astermark J, Benson G, Castaman G, et al. (2019) Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: Phase 2 trial results. Blood.
- Gu JM, Zhao XY, Schwarz T, Schuhmacher J, Baumann A, et al. (2017) Mechanistic Modeling of the Pharmacodynamic and Pharmacokinetic Relationship of Tissue Factor Pathway Inhibitor-Neutralizing Antibody (BAY 1093884) in Cynomolgus Monkeys. The AAPS Journal 19: 1186-1195.
- Sehgal A, Barros S, Ivanciu L, Cooley B, Qin J, et al. (2015) An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nature Medicine 21: 492-497.
- Polderdijk SG, Adams TE, Ivanciu L, Camire RM, Baglin TP, et al. (2017) Design and characterization of an APC-specific serpin for the treatment of hemophilia. Blood 129: 105-113.
- Rezaie AR, Manithody C, Yang L (2005) Identification of factor Xa residues critical for interaction with protein Z-dependent protease inhibitor: both active site and exosite interactions are required for inhibition. The Journal of Biological Chemistry 280: 32722-32728.
- Huang X, Dementiev A, Olson ST, Gettins PG (2010) Basis for the specificity and activation of the serpin protein Z-dependent proteinase inhibitor (ZPI) as an inhibitor of membrane-associated factor Xa. The Journal of Biological Chemistry 285: 20399-20409.
- Han X, Fiehler R, Broze GJ (2000) Characterization of the protein Z-dependent protease inhibitor. Blood 96: 3049-3055.
- Rezaie AR (2001) Prothrombin protects factor Xa in the prothrombinase complex from inhibition by the heparin-antithrombin complex. Blood 97: 2308-2313.
- Wood JP, Petersen HH, Yu B, Wu X, Hilden I, et al. (2017) TFPIalpha interacts with FVa and FXa to inhibit prothrombinase during the initiation of coagulation. Blood Advances 1: 2692-2702.
- Sierko E, Wojtukiewicz MZ, Zimnoch L, Ostrowska-Cichocka K, Tokajuk P, et al. (2012) Protein Z/protein Z-dependent protease inhibitor system in human non-small-cell lung cancer tissue. Thrombosis Research 129: e92-e96.
- Ferrigno D, Buccheri G, Ricca I (2001) Prognostic significance of blood coagulation tests in lung cancer. The European Respiratory Journal 17: 667-73.
- Levi M (2014) Cancer-related coagulopathies. Thrombosis Research 2: S70-S75.
- Cui J, Eitzman DT, Westrick RJ, Christie PD, Xu ZJ, et al. (2000) Spontaneous thrombosis in mice carrying the factor V Leiden mutation. Blood 96: 4222-4226.
- Hille ET, Westendorp RG, Vandenbroucke JP, Rosendaal FR (1997) Mortality and causes of death in families with the factor V Leiden mutation (resistance to activated protein C). Blood 89: 1963-1967.
- Sofi F, Cesari F, Abbate R, Gensini GF, Broze G, et al. (2010) A meta-analysis of potential risks of low levels of protein Z for diseases related to vascular thrombosis. Thrombosis and Haemostasis 103: 749-756.
- Martinelli I, Razzari C, Biguzzi E, Bucciarelli P, Mannucci PM (2005) Low levels of protein Z and the risk of venous thromboembolism. Journal of thrombosis and haemostasis: JTH 3: 2817-2819.
- Kurnik K, Kreuz W, Horneff S, During C, Schobess R, et al. (2007) Effects of the factor V G1691A mutation and the factor II G20210A variant on the clinical expression of severe hemophilia A in children--results of a multicenter studys. Haematologica 92: 982-9875.
- Shetty S, Vora S, Kulkarni B, Mota L, Vijapurkar M, et al. (2007) Contribution of natural anticoagulant and fibrinolytic factors in modulating the clinical severity of haemophilia patients. British Journal of Haematology 138: 541-544.
- Ghosh K, Shetty S, Mohanty D (2001) Milder clinical presentation of haemophilia A with severe deficiency of factor VIII as measured by one-stage assay. Haemophilia 7: 9-12.
- van Dijk K, van der Bom JG, Fischer K, Grobbee DE (2004) Do prothrombotic factors influence clinical phenotype of severe haemophilia? A review of the literature. Thrombosis and Haemostasis 92: 305-310.
- Franchini M, Lippi G (2010) Factor V Leiden and hemophilia. Thrombosis Research 125: 119-23.
- van 't Veer C, Golden NJ, Kalafatis M, Simioni P, Bertina RM, et al. (1997) An in vitro analysis of the combination of hemophilia A and factor V(LEIDEN). Blood 90: 3067-3072.
- Bos MH, Meijerman DW, van der Zwaan C, Mertens K (2005) Does activated protein C-resistant factor V contribute to thrombin generation in hemophilic plasma? Journal of Thrombosis and Haemostasis 3: 522-530.
- Schlachterman A, Schuettrumpf J, Liu JH, Furlan Freguia C, et al. (2005) Factor V Leiden improves in vivo hemostasis in murine hemophilia models. Journal of Thrombosis and Haemostasis 3: 2730-2737.
- Huang X (2019) Engineering a protein Z-dependent protease inhibitor (ZPI) mutant as a novel antagonist of ZPI anticoagulant function for hemophilia treatment. Journal of Thrombosis and Haemostasis 17: 1655-1660.
- Girard TJ, Lasky NM, Grunz K, Broze GJ (2019) Suppressing protein Z-dependent inhibition of factor Xa improves coagulation in hemophilia A. Journal of Thrombosis and Haemostasis 17: 149-156.
- Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, et al. (2013) Guidelines for the management of hemophilia. Haemophilia 19: e1-47.
- Wei Z, Yan Y, Carrell RW, Zhou A (2009) Crystal structure of protein Z-dependent inhibitor complex shows how protein Z functions as a cofactor in the membrane inhibition of factor X. Blood 114: 3662 -3667.
- Huang X, Zhou J, Zhou A, Olson ST (2015) Thermodynamic and kinetic characterization of the protein Z- dependent protease inhibitor (ZPI)-protein Z interaction reveals an unexpected role for ZPI Lys-239. The Journal of Biological Chemistry 290: 9906-9918.
- Huang X, Yan Y, Tu Y, Gatti J, Broze GJ, et al. (2012) Structural basis for catalytic activation of protein Z-dependent protease inhibitor (ZPI) by protein Z. Blood 120: 1726-1733.
- Almawi WY, Al-Shaikh FS, Melemedjian OK, Almawi AW (2013) Protein Z, an anticoagulant protein with expanding role in reproductive biology. Reproduction 146: R73-R80.
- Beerli RR, Bauer M, Fritzer A, Rosen LB, Buser RB, et al. (2014) Mining the human autoantibody repertoire: isolation of potent IL17A-neutralizing monoclonal antibodies from a patient with thymoma. mAbs 6: 1608-1620.
- Mosnier LO, Zlokovic BV, Griffin JH (2007) The cytoprotective protein C pathway. Blood 109: 3161-3172.
- Levy JH, Sniecinski RM, Welsby IJ, Levi M (2016) Antithrombin: anti-inflammatory properties and clinical applications. Thrombosis and Haemostasis 115: 712-728.
- Vasse M, Denoyelle C, Legrand E, Vannier JP, Soria C, et al. (2002) Weak regulation of protein Z biosynthesis by inflammatory cytokines. Thrombosis and Haemostasis 87: 350-351.
- Huang ZF, Higuchi D, Lasky N, Broze GJ (1997) Tissue factor pathway inhibitor gene disruption produces intrauterine lethality in mice. Blood 90: 944-951.
- Ishiguro K, Kojima T, Kadomatsu K, Nakayama Y, Takagi A, et al. (2000) Complete antithrombin deficiency in mice results in embryonic lethality. The Journal of Clinical Investigation 106: 873-878.
- Jalbert LR, Rosen ED, Moons L, Chan JC, Carmeliet P, et al. (1998) Inactivation of the gene for anticoagulant protein C causes lethal perinatal consumptive coagulopathy in mice. The Journal of Clinical Investigation 102: 1481-1488.
- Maroney SA, Cooley BC, Ferrel JP, Bonesho CE, Nielsen LV, et al. (2012) Absence of hematopoietic tissue factor pathway inhibitor mitigates bleeding in mice with hemophilia. Proceedings of the National Academy of Sciences of the United States of America 109: 3927-3931.
- Bolliger D, Szlam F, Suzuki N, Matsushita T, Tanaka KA, et al. (2010) Heterozygous antithrombin deficiency improves in vivo haemostasis in factor VIII-deficient mice. Thrombosis and Haemostasis 103: 1233-1338.
- Gupta A, Patibandla S (2019) Protein C Deficiency. StatPearls. Treasure Island (FL), 2019.
- Bravo-Perez C, Vicente V, Corral J (2019) Management of antithrombin deficiency: an update for clinicians. Expert Review of Hematology 12: 397-405.
- de Serres F, Blanco I (2014) Role of alpha-1 antitrypsin in human health and disease. Journal of Internal Medicine 276: 311-335.
- Bolton-Maggs PH, Pasi KJ (2003) Haemophilias A and B. Lancet 361: 1801-1809.
- Ragni MV (2015) Targeting antithrombin to treat hemophilia. The New England journal of Medicine 373: 389-391.
- George LA (2017) Hemophilia gene therapy comes of age. Blood Advances 1: 2591-2599.