Abstract
Gastrointestinal Stromal Tumors (GISTs) are rare sarcoma, yet stand as the predominant formin the intestinal tract. The majority of GISTs harbor activating mutations in KIT or PDGFRα, tyrosine kinase receptors. The mutation status upon diagnosis is essential to makemedical decisions forresectable or metastatic GIST patients, although surgery has been the mainstay of treatment in the early stage of the disease. The discovery and improvement of Tyrosine Kinase inhibitors (TKIs) against the KIT or PDGFRα have entailed targeted therapies based on specific molecular alterations. However, despite the clinical benefits of the first generation of TKI, imatinib, most GIST patients experience disease progression within 2–3 years of treatment. The 2nd and 3rd line TKIs include sunitinib and regorafenib, which have shown some efficacies in these imatinib resistant populations. This article aims to review the “gain of function”KIT or PDGFRα gene mutations by exons in the advanced GIST patients and the corresponding targeted therapies of these mutations.
Keywords
gastrointestinal stromal tumors, c-Kit,PDGFRα, targeted therapy
Overview of GIST
Gastrointestinal Stromal Tumors (GISTs) are and only account for 1-2% of gastrointestinal cancer [1]. However, it is the most common malignant mesenchymal subepithelial tumor. The GISTs are most found in the stomach and small intestine, but they can also occur in other GI and extra-GI sites [2-4]. GISTs are derived from mesenchymal stem cells known as Cajal’s interstitial cells, which has a function of coordinating gut motility [5], across the gastrointestinal tract. The annual incidence of GIST is approximately 10 per 1 million people and generally has an equal incidence rate in men and women. The disease typically affects middle-age to older patients with an average age of onset of 60 to 65 years old [2].
For those with early stage disease, surgical resection has been the main stay of treatment. However, recurrence and metastatic disease are common in high risk tumors. Traditional chemotherapy and radiotherapy are not effective on GISTs. A break through was made when c-Kit mutations were identified as the oncogenic driver for the development of GISTs [5]. Subsequently development of Imatinib, a multi-tyrosine kinase inhibitor, changed the clinical course of the disease [6,7].
Clinical Case
A 58-year-old female noticed increasing abdominal distension and girth over a 6-month period. The patient’s primary care physician ordered a CT scan of the abdomen that showed a 16cm abdominal mass on 2/16/2019. Further evaluation with a PET scan on 3/10/2019 confirmed the FDG avid mass (Figure 1). Biopsy of the mass revealed a GIST, with immuno histochemistry staining indicating positive for CD117 that is, the KIT protein. Due to an insufficient quantity of the biopsy specimen, sequencing analysis was not performed. She was treated with 400mg of imatinib daily for 3 months with the intention to downstage the tumor. However, the tumor did not respond to the therapy. On 7/24/2019, she underwent on block surgical resection with partial gastrectomy, partial duodenum resection, and colon resection. All the surgical margins were negative. The final pathology showed a T3N0M0 GIST, 0 mitosis per 50 high power field. Molecular analysis revealed the presence ofa Platelet-Derived Growth Factor Receptor Alpha (PDGFRA) exon 18 mutation present (D842V). Due to this rare and imatinib insensitive mutation, the patient did not receive adjuvant imatinib. She has been followed in clinic with periotic CT scans, history and physical examinations. She has been four years out from her surgery and has no evidence of disease.
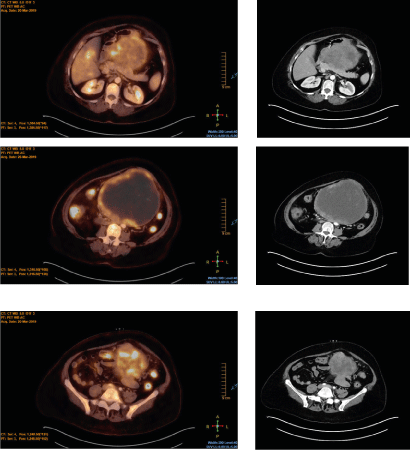
Figure 1. PET scan confirms FDG avid mass in a patient with
Common c-Kit mutations in GIST
Receptor tyrosine kinases play an important role in many cellular processes, and their dysregulation leads to diseases, most importantly cancer [8]. One such receptor tyrosine kinase is c-Kit (aka stem cell growth factor receptor), a type-III receptor tyrosine kinase, that is involved in various intracellular signaling pathways.c-Kit is a well-known cell surface receptor that binds to its physiological ligand, Stem Cell Factor (SCF), also known as c-Kit ligand, leading to several physiological functions. Physiologically, the activation of c-Kit occurs via binding of the SCF, leading to the dimerization of the receptor and the subsequent conformational changes, which evict the inhibitory Juxta Membrane (JM) domain from the split kinase domains. Activation of the kinasedomains requires a conformational change in the activation loop (A-loop), which enables the kinase domains to bind ATP and phosphorylatetarget substrates.This binding activates multiple downstream effectors including but not limited to Ras/MAP kinases, Src family kinases, p85 subunit of PI3 Kinase and phospholipase C-gamma, depending on the cell types [9,10].
Approximately 80% of GISTs exhibit KIT gene mutations, leading to constitutive activation of the KIT receptor and downstream signaling pathways that stimulate cell survival, growth, and proliferation. These “gain-of-function” mutations in the KIT gene in GISTs were identified in different exons of the gene and include point mutations, deletions, or insertions [5]. Therefore, the KIT gene has been considered a primary factor in the tumorigenesis of GISTs.
The KIT gene comprises of 21 exons: Exon 1 encodes the 5′-untranslated region and the signal peptide; Exons 2-9 encode the extracellular domain; Exon 10 encodes the transmembrane domain and the remaining exons encode the intracellular domain [11]. The extracellular region consists of five immunoglobulin (Ig)-like domains involved in ligand binding and receptor dimerization, and the transmembrane domain allows KIT anchoring in the plasma membrane. The cytoplasmic region is composed of a tyrosine kinase domain split in two by an interkinase domain and it is responsible for SCF/KIT signaling. KIT functional domains include the five extracellular immunoglobulin like domains (D1–5) responsible for ligand binding (D1-D3) and receptor dimerization (D4-D5), JM domain, tyrosine kinase 1 (TK1) domain, tyrosine kinase 2 (TK2) domain and activation loop (A-loop) [11] (Figure 2). Interestingly, mutations in different exons determines the sensitivity to Imatinib therapy as discussed below.
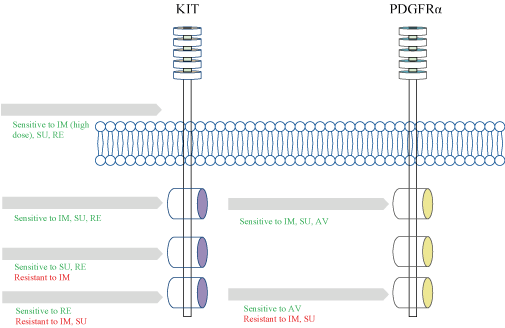
Figure 2. Diagram of the sensitivity of KIT and PDGFRA activating mutations to TKIs. IM = imatinib, SU = sunitinib, RE = regorafenib, AV= avapritinib
KIT Exon 11 mutation
Mutations in Exon 11 encodes the JM domain which sterically blocks the ATP-binding pocket and represents a half to two thirds of the KIT activating mutation in GIST [12]. Roughly 74.5% of all Exon 11 mutations involve codons 557 and/or 558. Both codons are associated with higher mitotic rate and worse prognosis, particularly double deletions [13]. Exon 11 mutations are the most sensitive to the Tyrosine Kinase Inhibitors (TKI) imatinib (90%) compared to its rarer counterparts. Patients with unresectable KIT exon 11 GISTs are typically given 400 mg of daily imatinib [14,15]. The response rate to imatinib is approximately 83.5% [16]. c-Kit exon 11 mutation in response to imatinib has the longest progression-free and overall survival than other exon mutations [16].
KIT Exon 9 mutation
Alterations in KIT Exon 9 coding for D1-5 was detected in 12-15% of GIST cases [12]. Clinically, approximately 85% of Exon 9-GISTs arise in extra-gastric sites, with a high predilection for the small intestine as 20–25% of all intestinal GISTs presented an Exon 9 mutation. Compared to most Exon 11-GISTs, Exon 9-GISTs have been associated with a more aggressive clinical behavior, and they have been reported to metastasize, significantly, more often to the peritoneum than to the liver, when compared to KIT/PGFRA wild-type and Exon-11-GISTs [17]. Exon 9 mutation is generally less sensitive to standard 400mg daily dose of imatinib. The response rate is about 47.8% comparing to 83.5% of exon 11 [16]. However, Gronchi and colleague showed increasing imatinib dose to 800 mg daily has clinical benefits to the patients with Exon-9-GISTs in Progression-Free-Survival (PFS) [18]. Although 400mg dose imatinib is the standard of care for adjuvant therapy, it is perceivable that 800mg dose should be considered in patients with exon 9 mutation.
KIT Exon 13, 14, 17, 18 mutations
Mutations in Exon 13 and 14 coding for TK1 domain and mutations in Exon 17 and 18 coding for A-loop are rare as they commonly occur as secondary KIT mutations. This results in outgrowth of heterogeneous subclones and subsequent treatment resistance [12,19]. Although the number of patients with those mutations is small, exon 13 and 17 are still considered responsive to imatinib.
The discovery of oncogenic KIT activation as a central mechanism of GIST pathogenesis suggested that inhibiting or blocking KIT signaling might be the milestone in the targeted therapy of GISTs. Indeed, imatinib mesylate inhibits KIT kinase activity, serving as the front-line drug for the treatment of unresectable and advanced GISTs, which achieves a partial response or stable disease in about 80% of patients with metastatic GIST. KIT mutation status has a significant impact on the treatment response, emerging in recent years as a leading paradigm for genotype-driven targeted therapy. The KIT mutations in different regions can affect the response to targeted therapy, and provide guidance for choosing an appropriate agent with optimal dosage. For example, tumors with KIT mutations in Exon 11 mutations are more sensitive to imatinib than those with Exon 9 mutations.Thus, it is important to obtain a molecular analysis of tumor specimen before initiating therapy. Our case confirmed that next generation sequencing should be integrated into our daily practice.
In 2006, sunitinib malate was approved by the FDA for the treatment of GIST after disease progression on (or intolerance to) imatinib, with a dose of 50 mg, 4 weeks on and 2 weeks off treatment. The continuous daily dosing of sunitinib at 37.5 mg daily is an active alternative dosing strategy with favorable safety [20,21]. In GIST patients progressing on sunitinib, regorafenib serves as a third line treatment [22]. Heterogeneity of KIT secondary mutations is the main reason for disease progression to KIT inhibitors in imatinib-resistant GIST patients. Preclinical study showed that the therapeutic combinations of TKIs with complementary activity against resistant mutations may be useful to suppress growth of polyclonal imatinib-resistance in GIST [19]. Of the 14 TKI-resistant GIST patients enrolled in a phase Ib trial that investigated treatment with three days of sunitinib followed by four days of regorafenib, 4 subjects achieved stable disease as the best response, the median progression-free survival was 1.9 months,and the median overall survival was 10.8 months. There were no unexpected toxicities resulted from this combinational therapy [23].
PDGFR mutations in GIST
Approximately 80% of GISTs are driven by activating oncogenic mutations in receptor kinase KIT while 5-10% by Platelet-Derived Growth Factor Receptoralpha (PDGFRA) [24]. PDGFRA is also a tyrosine kinase receptor, and phosphorylation substrates trigger activation of downstream pathways such as RAS-RAF-MEK-ERK (proliferation) and the PI3K-AKT-mTOR (survival) pathway. The alterations in PDGFRA are found explicitly in GIST [25] and are mutually exclusive of c-Kit mutation [26] (Figure 2). Although the downstream signaling pathways of KIT and PDGFRA mutant GIST were thought to be similar, gene expression profiling has shown that each classof mutations display differential expression of downstream signaling pathways. In an analysis of 26 GISTs, there were higher levels of AKT/PI3K pathway genes in KIT-mutated GISTs, while higher levels of genes associated with T-cell receptor signaling were seen in PDGFRA-mutated GISTs [27].
PDGFRA Exon 18 mutation
The most common PDGFRA mutation is Exon 18 (TK2 domain) D842V, a single nucleotide substitution 2664A→T leading to D842V activating mutation [28]. This mutation led to distortion of the kinase activation loop causing an altered protein conformation which favors the active structure [29]. This results in imatinib insensitivity in D842V mutants as imatinib can only bind to the inactive form of PDGFRA. Additionally, this mutation is also sunitinib resistance. As demonstrated in our case, imatinib is in effective in PEGFR D842V mutation. Based on the preliminary results of the Phase I NAVIGATOR study, avapritinib is the current international standard of care for PDGFRA Exon 18 D842V mutations [30]. In this study, 49 of 56 advanced GIST patients (88%) had an overall response; Progression-free survival was 100% at 3 months, 94%at 6 months, and 81% at 12 months; Overall survival was estimated to be 100% at 6 months, 91% at 12 months, and 81% at 24 months [30].
PDGFRA Exon 12 mutation
The second most common activating substitution of PDGFRA is on Exon 12, 1821T→A causing the V561D mutation (less than 1% of GISTs).
Other rare mutations including substitutions, deletions, duplications and insertions that have been reported and are physically close to exon 18 D842V or exon 12 V561D. However, due to their very low incidence, little is known about the specific behavior of these mutations [31,32]. PDGFRA mutants other than Exon 18 D842 mutants have been found to be imatinib sensitive [32].
The majority of the patients with PDGFR A have a gastric primary [28]. PDGFRA mutant GISTs are significantly more often very low/low risk comparedto those with KIT mutants (49 vs 26%), more often had tumors in the stomach (91 vs 45%) and more frequently had <5 mitoses per 50 high power field compared with KIT mutant GISTs [33]. Patients with tumors harboring PDGFRA mutations have a significantly better disease-free survival compared with those with tumors harboring KIT Exon 9 and 11 mutations [34].
Conclusion
In this report, we described a unique case of GIST, which presented a PDGFRA Exon 18 mutation (D842V) and showed no response to imatinib.The GIST treatment landscape is evolving and driven by further understanding of the disease’s molecular pathogenesis. Identification of molecular alterations of KIT and PDGFA not only provides the treatment options to the current patients with GIST but also the opportunity to develop the next generation of the targeted therapy.
References
- Beltran MA, Cruces KS (2007) Primary tumors of jejunum and ileum as a cause of intestinal obstruction: a case control study. Int J Surg 5: 183-191. [Crossref]
- Søreide K, Sandvik OM, Søreide JA, Giljaca V, Jureckova A, et al. (2016) Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol 40: 39-46. [Crossref]
- Miettinen M, Lasota J (2006) Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 23: 70-83. [Crossref]
- Machado-Aranda D, Malamet M, Chang YJ, Jacobs MJ, Ferguson L, et al. (2009) Prevalence and management of gastrointestinal stromal tumors. Am Surg 75: 55-60. [Crossref]
- Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, et al. (1998) Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279: 577-580.[Crossref]
- Wu PC, Langerman A, Ryan CW, Hart J, Swiger S, et al. (2003) Surgical treatment of gastrointestinal stromal tumors in the imatinib (STI-571) era. Surgery 134: 656-665. [Crossref]
- Corbin KS, Kindler HL, Liauw SL (2014) Considering the role of radiation therapy for gastrointestinal stromal tumor. Onco Targets Ther 7: 713-718. [Crossref]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, et al. (2013) Cancer genome landscapes. Science 339: 1546-1558. [Crossref]
- Gupta A, Ma S, Che K, Pobbati AV, Rubin BP (2021) Inhibition of PI3K and MAPK pathways along with KIT inhibitors as a strategy to overcome drug resistance in gastrointestinal stromal tumors. PLoS One 16: e0252689. [Crossref]
- Lennartsson J, Rönnstrand L (2012) Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev 92: 1619-1649. [Crossref]
- Chu TY, Besmer P (1995) The Genomic Structure of the Proto-Oncogene c-kit Encoded at the Murine White Spotting Locus. J Biomed Sci 2: 36-45. [Crossref]
- Hemming ML, Heinrich MC, Bauer S, George S (2018) Translational insights into gastrointestinal stromal tumor and current clinical advances. Ann Oncol 29: 2037-2045. [Crossref]
- Kontogianni-Katsarou K, Dimitriadis E, Lariou C, Kairi-Vassilatou E, Pandis N, et al. (2008) KIT exon 11 codon 557/558 deletion/insertion mutations define a subset of gastrointestinal stromal tumors with malignant potential. World J Gastroenterol 14: 1891-1897. [Crossref]
- (MetaGIST) GSTM-AG (2010) Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol 28: 1247-1253. [Crossref]
- Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, et al. (2009) Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 373: 1097-1104. [Crossref]
- Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, et al. (2003) Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 21: 4342-4349. [Crossref]
- Napolitano A, Thway K, Smith MJ, Huang PH, Jones RL (2022) KIT Exon 9-Mutated Gastrointestinal Stromal Tumours: Biology and Treatment. Chemotherapy 67: 81-90. [Crossref]
- Gronchi A, Blay JY, Trent JC (2010) The role of high-dose imatinib in the management of patients with gastrointestinal stromal tumor. Cancer 116: 1847-1858. [Crossref]
- Serrano C, Mariño-Enríquez A, Tao DL, Ketzer J, Eilers G, et al. (2019) Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br J Cancer 120: 612-620. [Crossref]
- Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, et al. (2006) Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368: 1329-1338. [Crossref]
- George S, Blay JY, Casali PG, Le Cesne A, Stephenson P, et al. (2009) Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer 45: 1959-1968. [Crossref]
- Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, et al. (2013) Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381: 295-302. [Crossref]
- Serrano C, Leal A, Kuang Y, Morgan JA, Barysauskas CM, et al. (2019) Phase I Study of Rapid Alternation of Sunitinib and Regorafenib for the Treatment of Tyrosine Kinase Inhibitor Refractory Gastrointestinal Stromal Tumors. Clin Cancer Res 25: 7287-7293. [Crossref]
- Antonescu CR (2011) The GIST paradigm: lessons for other kinase-driven cancers. J Pathol 223: 251-261. [Crossref]
- Lasota J, Miettinen M (2008) Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology 53: 245-266. [Crossref]
- Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, et al. (2003) PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299: 708-710. [Crossref]
- Subramanian S, West RB, Corless CL, Ou W, Rubin BP, et al. (2004) Gastrointestinal stromal tumors (GISTs) with KIT and PDGFRA mutations have distinct gene expression profiles. Oncogene 23: 7780-7790. [Crossref]
- Lasota J, Dansonka-Mieszkowska A, Sobin LH, Miettinen M (2004) A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest 84: 874-883. [Crossref]
- Szucs Z, Thway K, Fisher C, Bulusu R, Constantinidou A, et al. (2017) Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol 13: 93-107. [Crossref]
- Heinrich MC, Jones RL, von Mehren M, Schöffski P, Serrano C, et al. (2020) Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol 21: 935-946. [Crossref]
- Cassier PA, Fumagalli E, Rutkowski P, Schöffski P, Van Glabbeke M, et al. (2012) Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res 18: 4458-4464. [Crossref]
- Corless CL, Barnett CM, Heinrich MC (2011) Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer 11: 865-878. [Crossref]
- Wozniak A, Rutkowski P, Piskorz A, Ciwoniuk M, Osuch C, et al. (2012) Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann Oncol 23: 353-360. [Crossref]
- Wozniak A, Rutkowski P, Schöffski P, Ray-Coquard I, Hostein I, et al. (2014) Tumor genotype is an independent prognostic factor in primary gastrointestinal stromal tumors of gastric origin: a european multicenter analysis based on Contica GIST. Clin Cancer Res 20: 6105-6116. [Crossref]