The aim of this review is to summarize the most recent information about Silver-Russell syndrome (SRS), a clinically and genetically heterogeneous imprinting disorder that causes prenatal and postnatal growth retardation. Particular attention was focused on several recommendations for clinical diagnosis and management of patients with SRS published by the Consensus Statement on SRS in 2017.
Clinical aspects include intrauterine and postnatal growth retardation with relative macrocephaly, a typical triangular face, body asymmetry and other less specific features.
Diagnosis is still challenging because of clinical diagnosis not often confirmed by molecular findings. Overlap exists between the care of children born small for gestational age and those with SRS. However, several recommendations are specific for SRS. Treatment goals can be achieved by a multidisciplinary team approach, but natural history of this disease should be studied by long-term follow up until adulthood.
growth retardation, Silver Russell syndrome, imprinting disorder, small for gestational age, short stature
Silver–Russell Syndrome (SRS) is a rare, clinically and genetically heterogeneous disorder associated with prenatal and postnatal growth retardation. Silver et al. [1] in 1953 and Russell [2] in 1954 independently described the syndrome for the very first time, reporting a subset of children with low birth weight, short stature, body asymmetry and characteristic facial features.
At birth, all patients with SRS are small for gestational age (SGA); however, children with SRS present other peculiar clinical features that can differentiate SRS from idiopathic intra-uterine growth retardation or SGA. Suggestive clinical characteristics of SRS in the newborn are: asymmetric gestational growth restriction with relative macrocephaly [defined as a head circumference at birth ≥1.5 SD score (SDS) above birth weight and/or length SDS], prominent forehead, body asymmetry and feeding difficulties [3-6].
The real frequency of the disease is unknown, but it is probably under diagnosed due to its different and heterogeneous features. These features are non-specific and can vary widely in severity. Furthermore, clinical signs are peculiar in infancy and early childhood but can become less evident in older children. The incidence of SRS globally ranges from 1:30,000 to 1:100,000 [7]. A recent retrospective study in Estonia estimated the minimum prevalence of SRS at birth as 1:15,886 [8].
Studies on SRS are challenging even in genetic assessment. An underlying molecular cause can be identified in around 60% of patients clinically diagnosed with SRS.
The majority of SRS cases are sporadic, but various ways of inheritance including recessive, dominant and X-linked have been suggested [9]. Several chromosomal aberrations have been associated with SRS, including chromosome 1, 2, 7, 8, 11, 12, 13, 14, 15, 16, 17, 18, 20, 21, 22 and X. However, some of these reported cases include SRS-like patients who do not match the criteria for diagnosing SRS. The most common underlying mechanisms are loss of methylation on chromosome 11p15 (11p15 LOM; seen in 30 to 60% of patients) and maternal uniparental disomy for chromosome 7 (UPD7; seen in ~5-10% of patients) [10].
Rarely, affected individuals with pathogenic variants in CDKN1C, IGF2, PLAG1, and HMGA2 have been described. However, approximately 40% of individuals who meet NH-CSS clinical criteria for SRS have negative molecular and/or cytogenetic testing [11].
A consensus meeting developed guidelines for the diagnosis and management of patients with SRS. This consensus is important to clarify the overlap in the clinical care of SGA individuals and those with SRS. Diagnosis and therapeutic approach can be specific in SRS individuals.
The Consensus Statement involved different academic societies: the COST Action BM1208 (European Network for Human Congenital Imprinting Disorders, http://www. imprinting-disorders.eu), European Society of Pediatric Endocrinology (ESPE), Pediatric Endocrine Society (PES), Asian Pacific Pediatric Endocrine Society (APPES) and Sociedad Latino-Americana de Endocrinología Pediátrica (SLEP).
The aim of this review is to focus on the new knowledge and implications about SRS. A comprehensive literature research was conducted using PubMed by the search terms “Silver Russell Syndrome”. Reviews on this topic were mainly considered. Furthermore, articles on genetic SRS implications and its molecular aspects, differential diagnosis and treatment were included in PubMed searches in order to have additional information.
Particular attention was focused on the first consensus statement on SRS published in 2017. Several recommendations were established; in this review only those with strong evidence will be mentioned.
Clinical characteristics
Silver-Russell Syndrome (SRS) is characterized by peculiar features: asymmetric gestational growth restriction resulting in SGA newborns with relative macrocephaly (head circumference ≥1.5 SD above birth weight and/or length), prominent forehead usually with frontal bossing and body asymmetry. SRS children show postnatal growth failure (< 2 SD at 24 months) and severe feeding difficulties in the first years of life; in addition, a typical face (triangular shaped face and micrognathia with narrow chin) has been described. The growth failure in SRS is proportionate with normal head growth. Growth charts for European children with SRS have been published: at the age of 2 years, most children with SRS remain >2 SD below the mean for length. The average adult height in untreated individuals is ~3.1 ± 1.4 SD below the mean [12].
In two European studies on untreated adults with SRS, height ranged from 3.7 to 3.5 SD below the mean for males and 4.2 to 2.5 SD below the mean for females [13].
SRS clinical diagnosis can be reached by using the Netchine-Harbison Clinical Scoring System (NH-CSS; Table 1), described as the most sensitive of the compared diagnostic scoring systems [14,15].
Table 1: The Netchine-Harbison Clinical Scoring System for Clinical Diagnosis (NH-CSS) of Silver-Russell syndrome
Clinical Criteria |
Definition |
SGA (birth weight and/or birth length) |
£ -2 SDS for gestational age |
Postnatal growth failure |
Height at 24 ± 1 months £ -2 SDS or height £ -2 SDS below mid-parental target height |
Relative macrocephaly at birth |
Head circumference at birth ³1.5 SDS above birth weight and/or length SDS |
Protruding forehead* |
Forehead projecting beyond the facial plane on a side view as a toddler (1-3 years) |
Body asymmetry |
LLD of ³ 0.5 cm or arm asymmetry or LLD <0.5 cm with at least two other asymmetrical body parts (one non-face) |
Feeding difficulties and/or low BMI |
BMI £ -2 SDS at 24 months or current use of a feeding tube or cyproheptadine for appetite stimulation |
*Protruding forehead is equivalent to ‘prominent forehead’. LLD: Leg Length Discrepancy; SDS: SD score; SGA, Small for Gestational Age.
Clinical diagnosis is reached when an infant meet at least four of the clinical criteria, two of which must be relative macrocephaly at birth and frontal bossing.
Supportive clinical findings
SRS individuals may have additional supportive clinical findings (Table 2), as described below.
Table 2: Supportive clinical findings in Silver Russell Syndrome patients
Supportive clinical findings |
Features |
Craniofacial anomalies |
Pierre Robin sequence
Cleft palate
Down-turned corners of the mouth Micrognathia
High-arched palate
Dental and oral abnormalities |
Neurodevelopment issues |
Developmental delay
Learning difficulties |
Feeding disorders and hypoglycemia |
Poor appetite
Oral motor problems |
Gastrointestinal disorders |
Gastroesophageal reflux disease
Esophagitis
Failure to thrive |
Muscle-Skeletal abnormalities |
Hemi-hypotrophy with limb length asymmetry Fifth-finger clinodactyly and/or brachydactyly
Scoliosis or kyphosis
Shoulder dimples
Diminished muscle mass
Hypoplastic elbow joints |
Hormonal abnormalities |
GH deficiency
Premature adrenarche
Early puberty
Insulin resistance |
Genitourinary problems |
Hypospadias and cryptorchidism
Mayer-Rokitansky-Kuster-Hauser syndrome |
Heart defects |
Rare but reported congenital heart defects |
Craniofacial anomalies: Craniofacial anomalies are common. Pierre Robin sequence and cleft palate are present in some individuals. Cleft palate or bifid uvula were assessed in 7% of those with 11p15.5 methylation defects and in no individuals with maternal UPD7 [16]. Obstructive apnea can occur in those individuals with Pierre Robin sequence. Other typical face characteristics are also present, such as down-turned corners of the mouth, micrognathia, high-arched palate, dental and oral abnormalities [17]. The most common orofacial manifestations are overbite and dental crowding [18].
Neurodevelopment issues: SRS children seem to be at increased risk for developmental delay (both motor and cognitive) and learning difficulties. In a review of a large cohort of children with SRS with either 11p15 methylation defects or maternal UPD7, developmental delay was observed in 34% of individuals, the majority of whom had mild delays. Developmental delays were more commonly seen in those with maternal UPD7 than in those with 11p15 methylation defects (65% vs 20%). Speech delays were common in both groups.
Feeding disorders and hypoglycemia: SRS individuals often have poor appetite and feeding disorders including oral motor problems [19]. The risk for hypoglycemia is high, especially associated with any prolonged fasting [20]. Factors that may favor hypoglycemia in SRS children are: reduced body mass index; reduced caloric intake, often secondary to poor appetite and feeding; in addition, growth hormone (GH) deficiency may be present [21].
Gastrointestinal disorders: Gastrointestinal disorders are common including gastroesophageal reflux disease, esophagitis and failure to thrive. Gastrointestinal problems affect 77% of children with SRS, and 55% of children had severe gastroesophageal reflux [22].
Muscle-Skeletal abnormalities: Muscle-Skeletal abnormalities are often present, such as hemi-hypotrophy with limb length asymmetry, fifth-finger clinodactyly and/or brachydactyly, scoliosis or kyphosis (described in 21% of individuals; 18% required corrective surgery) [23], shoulder dimples, diminished muscle mass, hypoplastic elbow joints.
Hormonal abnormalities: Hormonal abnormalities are also frequent, such as premature adrenarche, early puberty and insulin resistance.
Genitourinary problems: Common anomalies are hypospadias and cryptorchidism in males [24]. Mayer-Rokitansky-Kuster-Hauser syndrome (associated with underdeveloped or absent vagina and uterus with normal appearance of the external genitalia) has been reported in females [25]. Renal anomalies are not common; however, horseshoe kidney and renal dysplasia have been observed.
Heart defects: Heart defects are rare, but have been reported in larger studies and smaller case series. The prevalence of heart defects may be as high as 5.5% [26].
Genetic and molecular aspects
Molecular basis of SRS is still not completely known. People normally inherit one copy of each chromosome from their mother and one copy from their father. For most genes, both copies are expressed; for some genes, however, only the copy inherited from a person's father (the paternal copy) or from a person's mother (the maternal copy) is expressed [27]. These parent-specific differences in gene expression are caused by a phenomenon called genomic imprinting. Imprinting disorders (IDs) are characterized by molecular alterations at the imprinted loci. Imprinted genes are found in clusters under coordinated control. This control is mediated by differentially methylated regions (DMRs). The epigenetic marks in these DMRs are acquired during gametogenesis, and normal embryo development is dependent on their maintenance after fertilization and during embryogenesis [28,29].
IDs are associated with changes in imprinting [30]. Molecular disorders in SRS often result from the abnormal regulation of certain genes that control growth. These genes are located in particular regions of chromosome 7 and chromosome 11. Both chromosome 7 and chromosome 11 contain groups of genes that normally undergo genomic imprinting; some of these genes are active only on the maternal copy of the chromosome while others are active only on the paternal copy. Abnormalities involving these genes appear to be responsible for many cases of SRS. Around half of SRS cases result from changes in a process called methylation on the short arm of chromosome 11 at position 15 (11p15). Methylation is a chemical reaction that attaches small molecules called methyl groups to certain segments of DNA. In genes that undergo genomic imprinting, methylation is one way that a gene's parent of origin is marked during the formation of egg and sperm cells. SRS has been associated with changes in methylation involving the H19 and IGF2 genes, which are located near one another at 11p15. These genes are involved in normal growth development. A loss of methylation disrupts the regulation of these genes, which leads to slow growth and other characteristic features of this syndrome [31,32].
In 10% of children with SRS is abnormalities on chromosome 7 have been observed. It happens when children inherit both copies of chromosome 7 from their mother instead of one copy from each parent. This phenomenon is called maternal unipaternal disomy (UPD). Maternal UPD causes people to have two active copies of some imprinted genes and no active copies of others. An imbalance in certain active paternal and maternal genes on chromosome 7 results in clinical signs of SRS.
In addition to these two main mechanisms, several rare genetic disorders have been described: copy number variation (CNV) within the 11p15.5 domain, mostly involving maternal duplications [33]; rare paternal deletions of enhancers in the telomeric domain, leading to lower levels of IGF2 expression [34]; gain-of-function CDKN1C mutations (identified in a familial case of SRS) [35]; loss-of-function IGF2 mutation, exome sequencing in a family including three patients with the SRS phenotype [36]; CNV outside the 11p15 region and maternal UPD of other chromosomes [37]; abnormalities of chromosome 14.
Furthermore, in about 40 % of people with SRS the cause of the condition is unknown.
Investigation and diagnosis
Clinical diagnosis is considered if a patient scores at least four of six criteria from the Netchine-Harbison Clinical Scoring System (NCHSS). However, clinical diagnosis of children with SRS has to be confirmed by molecular testing. Molecular confirmation is useful for the stratification into a specific molecular subgroup and, consequently, for an appropriate management. If the patient meets clinical criteria for diagnosis of SRS (NCHSS) or if the clinical suspicion is strong, molecular testing for 11p15 and UDP(7)mat is required. In positive cases, molecular SRS diagnosis is confirmed. In negative cases, differential diagnosis has to be considered assessing clinical features consistent with other syndromic diagnoses. If other diagnoses are excluded, additional molecular testing can be evaluated: CNV and/or 14q32 analysis followed by UDP(16)mat, UDP(20)mat or CDKN1C or IGF2 mutation analysis. If all molecular tests are normal and differential diagnoses have been ruled out, patients scoring at least four of six criteria, including both prominent forehead and relative macrocephaly should be diagnosed as clinical Silver–Russell syndrome (Figure 1).
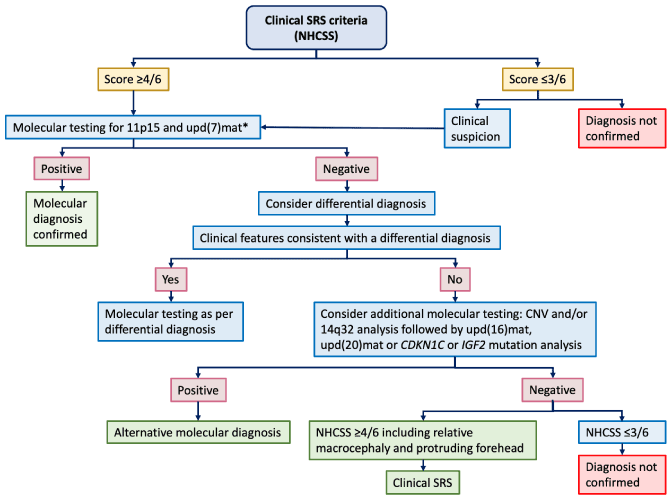
Figure 1: Flow chart for investigation and diagnosis of SRS proposed by Wakeling et al. in the first international consensus statement, 2017 (modified).
*Arrange CNV analysis before other investigations if patient has notable unexplained global developmental delay and/or intellectual disability and/or relative microcephaly.
CNV: Copy Number Variant; NH-CSS: Netchine-Harbison Clinical Scoring System; SRS, Silver–Russell Syndrome.
Genotype-phenotype correlations
Delineation of genotype-phenotype is challenging, because sometimes the molecular confirmation of clinical diagnosis is difficult. Furthermore, the presence of mosaicism in a subgroup of SRS patients allows the escape of molecular diagnosis on blood cells. Indeed, the SRS phenotype of carriers of the 11p15 epimutation is generally more severe and typical than that of UPD(7)mat carriers [38,39]. However, the phenotypic transition can change widely, therefore carriers of 11p15 epimutations and UPD(7)mat cannot be discriminated solely by clinical findings [40]. Bruce et al. [41] showed that, after distinguishing SRS individuals with extreme, moderate, normal H19 hypomethylation and maternal UPD7 (normal H19 methylation) by using methylation-sensitive restriction enzymes Hpall or Notl), children with extreme H19 hypomethylation were more likely to have severe skeletal manifestations (including greater limb asymmetry, syndactyly and scoliosis) than children with SRS with moderate hypomethylation and those with maternal UPD7. Hall [42] compared clinical features of children with SRS caused by 11p15.5 ICR1 IGF2/H19 methylation defects to those with maternal UPD7 and found that fifth-finger clinodactyly and congenital anomalies were more frequent in children with 11p 15.5 ICR1 hypomethylation than in those with maternal UPD7, whereas learning difficulties and speech disorders were more frequent in children with maternal UPD7 than in those with ICR1 hypomethylation.
SRS children with maternal UPD7 had more gain in height with GH therapy compared to children with 11p15.5 epimutations, possibly because children with 11p15.5 methylation abnormalities showed elevated levels of insulin-like growth factor I (IGF-1) and therefore a degree of IGF-1 resistance; children with SRS with maternal UPD7 responded to treatment similarly to other children who were small for gestational age.
Differential diagnosis
The differential diagnosis of children with short stature at birth includes syndromic diagnoses and chromosomal rearrangements. Specific clinical characteristics should suggest other diagnoses other than SRS. These include relative microcephaly (head circumference SDS below height and weight SDS), evident global developmental delay or intellectual disability (without a related explanation such as documented hypoglycemia), absence of severe feeding difficulties and/or the presence of additional congenital anomalies, facial dysmorphism or other features atypical of SRS. Disproportionate short stature is suggestive of skeletal dysplasia. SRS is generally sporadic, then a family history of growth failure might suggest an alternative underlying diagnosis [43].
Patients with features of SRS overlapping with osteogenesis imperfecta should have a skeletal survey in order to consider COL1A1/2 gene testing.
Management
Multidisciplinary care in a center of expertise in SRS is needed. The team should be composed of pediatric subspecialists such as an endocrinologist (coordinator), a gastroenterologist, a dietician, a clinical geneticist, a craniofacial team, an orthopedic surgeon, a neurologist, a speech and language therapist and a psychologist.
Early feeding and nutritional support
Neonates with SRS have body asymmetry with relative macrocephaly (length SDS below weight SDS); but after birth, because of feeding difficulties and gastrointestinal problems, weight SDS drops below the length SDS [44,45]. Length deficit become progressively more important with growing up. In SRS children combined factors as functional-structural gastrointestinal problems and feeding difficulties (poor appetite, oral motor problems) result in failure to thrive. Digestive problems or malnutrition occur in over 70% of patients with SRS, including severe gastroesophageal reflux in 55% after the age of one year and constipation, particularly after the age of two years. Weight gain could improve with cyproheptadine treatment [46,47].
The goals in nutritional support as established by the Consensus Statement on SRS in 2017 are: in the first years of life, nutritional repletion is needed avoiding rapid postnatal catch-up and its subsequent metabolic risk; early screening for gut dysmotility (gastro-esophageal reflux, delayed gastric emptying and constipation) in all children; diagnose and treat any oro-motor issue that affect oral intake of food; avoid enteral feeding by nasogastric or gastrostomy tube if the child is able to eat; in cases of extreme feeding difficulties consider enteral feeding by gastrostomy tube or low-profile trans-gastric jejunostomy as a last resort to protect against hypoglycemia and/or malnutrition.
Prevention of hypoglycemia
SRS children under the age of five have low muscle and liver mass, a disproportionately large brain-for-body size and feeding difficulties with increased risk of fasting hypoglycemia.
Prevention of hypoglycemia can be reached by: monitoring for ketonuria at home; developing a plan with the child’s local pediatrician and parents for rapid admission to hospital and intravenous dextrose treatment when the child is sick; glucagon is not recommended to correct hypoglycemia, because of poor glycogen stores and limited ability for gluconeogenesis; teaching parents how to recognize signs of hypoglycemia, measure ketones, determine the ‘safe fasting time’ for their child, prevent hypoglycemia using complex carbohydrates. In severe cases of fasting hypoglycemia, if other alternatives are not effective, early start of GH therapy (allowing muscle mass increase and gluconeogenesis) or placement of a gastrostomy tube or jejunostomy tube can be considered in order to support glucose sources.
Growth hormone treatment
Despite the lack of data in literature, SRS is often associated with an important reduction in adult height (around −3 SDS) [48]. GH treatment does not have a specific indication for SRS and is prescribed under the SGA indication [49]. Clinical trials of GH in short children born SGA paved the way for approval of the use of GH treatment by US FDA and the European Medicines Agency (EMA) in 2001 and 2003, respectively.
Overall, clinical trials of GH treatment in patients with SGA (in which patients with SRS were included) showed a satisfactory growth response and an increase in predicted adult height of 7-11 cm at pharmacological doses of GH [50-54]. The response to GH treatent in patients with SRS compared with non-SRS children born SGA is similar (mean total height gains of 1.30 SDS and 1.26 SDS, respectively); however, the final adult height reached by SRS patients seems to be lower (mean adult height −2.17 SDS versus −1.65 SDS for non-SRS children born SGA) [55]. Anyhow, it is necessary to undeline that the mean height at the start of GH treatment in patients with SRS was significantly lower than in those without SRS.
Age and height SDS at the start of GH treatment showed to be strong predictors of the short-term and long-term responses to GH treatment (both inversely related) [56,57]. Additional potential benefits of GH treatment are increases in appetite, lean body mass and muscle power, which can result in improved mobility [58,59]. Therefore, GH treatment is recommended from early childhood (age 2-4 years) in SRS children. Starting GH therapy below the age of 2 years can be considered in case of: severe fasting hypoglycemia; severe malnutrition despite nutritional support, which will lead to gastrostomy if no improvement is seen; and severe muscular hypotonia. Classic GH deficiency is neither a common nor a relevant cause of short stature in SRS, nor is it predictive of the response to GH treatment in children born SGA [60]. For most children with SRS, an increase in height velocity of more than 3 cm per year is the lower limit of an effective response range. A correct assessment of GH treatment can be achieved by monitoring circulating levels of IGF-1 and IGFBP-3 at least yearly. The 2017 Consensus Statement recommended to terminate GH therapy when height velocity is less than 2 cm per year over a 6-month period and bone age is more than 14 years in girls or 17 years in boys. If growth velocity is poor, re-evaluation of the underlying diagnosis, GH dose, IGF-1 response, adherence to therapy and other confounding systemic problems need to be investigated.
Bone age advancement and puberty
Data on natural history of bone age progression in patients with SRS are lacking. Early bone age delay is followed by rapid advancement at around 8-9 years of age [61] but sometimes in a much younger age. Onset of puberty is usually within the normal range (8-13 years in girls and 9-14 years in boys) [62] but at the younger end of the spectrum [63]. In some SRS individuals adrenarche can be early and aggressive in comparison with children born with non-SRS SGA (particularly in those with 11p15 LOM116). This early puberty further accelerates bone age maturation, which leads to an attenuated pubertal growth spurt and compromised adult height. A rapid increase in BMI might also exacerbate the tendency to early adrenarche and central puberty [64-66]. The 2017 Consensus Statement recommended to monitor for signs of premature adrenarche, early and accelerated central puberty, insulin resistance and anticipate acceleration of bone age, especially from mid childhood. It is convenient to consider personalized treatment with Gn-RH analogues for at least 2 years in children with evidence of central puberty (starting no later than age 12 years in girls and age 13 years in boys) to preserve adult height potential.
Long-term metabolic complications
Individuals born with a low birth weight have increased risk of developing metabolic complications as coronary heart disease [67-69], hypertension, dyslipidemia, obesity, insulin resistance (the metabolic syndrome) and development of type 2 diabetes mellitus [70]. Children born SGA that have rapid catch-up in weight are at particularly high risk [71-72]. Overall, GH therapy seems to have positive metabolic effects in children born SGA [73] increasing lean body mass, reducing fat mass, decreasing blood pressure and improving lipid profile [74,75]; however, data concerning with the aforementioned effects in SRS are lacking.
The goals in metabolic control are: avoid of excessive or rapid weight gain and increased insulin resistance; prevention of complications with collaboration of gastroenterologists, dieticians, neonatologists, pediatricians and primary health-care providers. Promoting a healthy diet and lifestyle with particular emphasis on protein calorie balance and regular exercise is a successful strategy.
Neurocognitive problems
Children with SRS are at risk of developing motor and speech delay. In particular those with UPD(7)mat can present verbal dyspraxia, learning difficulties, autistic spectrum disorder and myoclonus dystonia [76-78]. It is important to monitor neurocognitive development for early and appropriate interventions if necessary. In children with UPD(7)mat if signs of myoclonus dystonia, verbal dyspraxia, learning difficulties or autistic spectrum disorder appears it is necessary to refer to a pediatric neurologist. It is important to inform parents about increased risk of speech, oro-motor and learning disabilities [especially in those with UPD(7)mat].
Orthopaedic problems
SRS children can present relevant orthopedic problems (limb or body asymmetry, scoliosis, hip dysplasia and hand and/or foot anomalies). If necessary, refer to a pediatric orthopedic surgeon for correct management of these issues. Regular control of leg length asymmetry is important. The role of GH treatment on exacerbating scoliosis is still not clear, but before starting GH therapy it is recommended to refer to the orthopedic team and monitor while receiving GH.
Maxillofacial abnormalities
Craniofacial abnormalities are common in SRS children, resulting in a typical triangular-shaped face [79] with microdontia, absence of secondary teeth, small, pointed chin, overbite and velopharyngeal insufficiency [80-82]. It is important to refer to a maxillofacial team, including orthodontists, plastic surgeons and ear, nose and throat surgeons if problems may occur.
Many patients with SRS report sleep disorders with daytime fatigue [83]. Screening for symptoms of sleep disordered breathing (snoring, apneas, daytime fatigue, agitation) is necessary for evaluation of obstructive sleep apnea.
Other congenital abnormalities
Other anomalies have been described in association with SRS. Female patients can have hypoplasia or aplasia of the uterus and upper part of the vagina (Mayer-Rokitansky-Kuster-Hauser syndrome) [84]. Boys can present with genital abnormalities, including cryptorchidism and hypospadias. In addition, renal anomalies and congenital heart defects have also been reported. Thus, investigation of genital abnormalities in SRS children should be managed routinely.
Genetic counselling
Data about the risk of parents of children with clinically diagnosed SRS to have another child with SRS are lacking; however, the overall risk seems to be low. The offspring risk for individuals with clinically diagnosed SRS seems to be also low.
Obviously, the underlying molecular mechanism influences a correct genetic counselling. Recurrence risk is low in 11p15 LOM. Familial cases of SRS are rare and usually related to several underlying molecular mechanisms including: maternally inherited 11p15 duplication [85,86]; maternally inherited CDKN1C gain-of-function mutations [87]; and paternally inherited IGF2 loss-of-function mutations [88]. In these cases, the risk of recurrence could be around 50%. UPD(7)mat is associated with a low recurrence and offspring risk [89].
SRS and assisted reproduction
Assisted reproduction technologies (aRT) seem to be associated to an increased risk of imprinting disorder diseases (such as SRS, Beckwith-Wiedemann and Angelman syndromes) in newborns [90,91]. Imprinting mechanisms occurs in pre-implanted embryos; in this critical period of development, aRT procedures can influence genomic imprinting. Concurring mechanisms at the base of this hypothetic damage remain unknown, however, growth factors used during aRT procedures can possibly have a role on the methylation of the embryo genome. Some studies evidenced an increased risk of SRS in children born from aRT procedures, particularly in those with IcR1 11p15 methylation defects. Several reports focused on the links between IDs and the different aRT procedures, but these studies were underpowered. In addition, the absence of a well delineated aRT protocol from country to country, the limitation of sample size and the lack of data over all confounding factors that may act in such a complicated procedure, make this association difficult to be defined. In conclusion, this relationship remains uncertain. Finally, parents who are planning to use aRT procedures should be informed about the possible increased risk of imprinting disorders. For the same reason, neonatologists and neonatal nurse should monitor aRT infants according to their elevated risk on developing imprinting disorders.
Prenatal testing
In the last years requests for the prenatal testing of imprinting disorders is increasing, especially in cases of early intrauterine growth retardation. Data for suspected prenatal cases of SRS from several expert centers have recently been reported [92]. These data relate with both false-positive and false-negative results, demonstrating low sensitivity and specificity of these tests on detecting imprinting disorders. Prenatal test is not accurate, probably because of inappropriate sampling times and mosaicism that can cause a false-negative testing result. Furthermore, there is no consensus on the target methylated CpGs and DMRs in ID testing and, because of the heterogeneity and complexity of the molecular findings in the 11p15-associated IDs, unusual molecular alterations may elude identification.
Indication for prenatal molecular testing should, therefore, be discussed carefully by the parents, physician and molecular geneticist. In particular, any decisions regarding the outcome of the pregnancy to be taken on the basis of a positive or negative result of the molecular test should be considered beforehand and the parents and the physician should be aware of the possibility of false-positive or false-negative results [93].
SRS is a challenging disease for families of affected children and for physicians. Modern medicine is developing new diagnostic and management insights. The First International Consensus Statement on SRS published guidelines for the diagnosis and management of SRS. This is a clear sign that the attention on this disease is vivid. In this review we tried to summarize all the 72 recommendations that experts on SRS have elaborated. The most challenging issues that physicians should face on with SRS patients are: severe prenatal and postnatal growth failure with no catch-up, feeding difficulties, metabolic disorders as recurrent hypoglycemia and insulin resistance, precocious puberty, body asymmetry and the potential for other congenital anomalies. Follow up of SRS patients should be managed by a multidisciplinary team.
Since data on molecular aspects are still lacking, a molecular-subtype-guided management it’s not completely possible. Further studies will contribute to clarify the unsolved questions in order to improve long-term prognosis of children with SRS.
MR and FC contributed equally to writing the manuscript.
- Russell A (1954) A syndrome of intra-uterine dwarfism recognizable at birth with cranio-facial dysostosis, disproportionately short arms, and other anomalies (5 examples). Proc R Soc Med 47: 1040–1044. [Crossref]
- Silver HK, Kiyasu W, George J, Deamer WC (1953) Syndrome of congenital hemihypertrophy, shortness of stature, and elevated urinary gonadotropins. Pediatrics 12: 368–376. [Crossref]
- Wollmann HA, Kirchner T, Enders H, Preece MA, Ranke MB (1995) Growth and symptoms in Silver– Russell syndrome: review on the basis of 386 patients. Eur J Pediatr 154: 958–968. [Crossref]
- Netchine I, Rossignol S, Dufourg MN, Azzi S, Rousseau A, et al. (2007) 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell–Silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J Clin Endocrinol Metab 92: 3148–3154. [Crossref]
- Price SM, Stanhope R, Garrett C, Preece MA, Trembath RC (1999) The spectrum of Silver–Russell syndrome: a clinical and molecular genetic study and new diagnostic criteria. J Med Genet 36: 837–842
- Wakeling EL (2011) Silver–Russell syndrome. Arch Dis Child 96: 1156–1161. [Crossref]
- Toutain A (2007) Silver–Russell syndrome. Orphanet
- Yakoreva M, Kahre T, Žordania R, Reinson K, Teek R, et al. (2019) A retrospective analysis of the prevalence of imprinting disorders in Estonia From 1998 to 2016. Eur J Hum Genet 27: 1649-1658. [Crossref]
- Bliek J, Terhal P, van den Bogaard MJ, Maas S, Hamel B, et al. (2006) Hypomethylation of the H19 gene causes not only Silver–Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am J Hum Genet 78: 604–614. [Crossref]
- Schönherr N, Meyer E, Eggermann K, Ranke MB, Wollmann HA, et al. (2006) (Epi)mutations in 11p15 significantly contribute to Silver–Russell syndrome: but are they generally involved in growth retardation? Eur J Med Genet 49: 414–418. [Crossref]
- Saal HM, Harbison MD, Netchine I (2002) Silver-Russell Syndrome. GeneReviews [Crossref]
- Wollmann HA, Kirchner T, Enders H, Preece MA, Ranke MB (1995) Growth and symptoms in Silver-Russell syndrome: review on the basis of 386 patients. Eur J Pediatr 154: 958–968. [Crossref]
- Binder G, Liebl M, Woelfle J, Eggermann T, Blumenstock G, Schweizer R (2013) Adult height and epigenotype in children with Silver-Russell syndrome treated with GH. Horm Res Paediatr 80: 193–200. [Crossref]
- Netchine I, Rossignol S, Dufourg MN, Azzi S, Rousseau A, et al. (2007) 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell-Silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J Clin Endocrinol Metab 92: 3148–54. [Crossref]
- Azzi S, Salem J, Thibaud N, Chantot-Bastaraud S, Lieber E, et al. (2015) A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver- Russell syndrome. J Med Genet 52: 446–53. [Crossref]
- Wakeling EL, Amero A, Alders M, Bliek J, Forsythe E, et al. (2010) Epigenotype-phenotype correlatinos in Silver-Russell syndrome. J Med Genet 47: 760–768. [Crossref]
- Orbak Z, Orbak R, Kara C, Kavrut F (2005) Differences in dental and bone maturation in regions with or without hemihypertrophy in two patients with Russell-Silver syndrome. J Pediatr Endocrinol Metab 18: 701–710. [Crossref]
- Hodge N, Evans CA, Simmons KE, Fadavi S, Viana G (2015) Occlusal Characteristics of Individuals with Growth Hormone Deficiency, Idiopathic Short Stature, and Russell-Silver Syndrome. J Dent Child (Chic) 82: 135–40. [Crossref]
- Fuke T, Mizuno S, Nagai T, Hasegawa T, Horikawa R, et al. (2013) Molecular and clinical studies in 138 Japanese patients with Silver-Russell syndrome. PLoS One 8: e60105. [Crossref]
- Wakeling EL, Brioude F, Lokulo-Sodipe O, O'Connell SM, Salem J, et al. (2017) Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol 13: 105–124. [Crossref]
- Azcona C, Stanhope R (2005) Hypoglycaemia and Russell-Silver syndrome. J Pediatr Endocrinol Metab 18: 663–670. [Crossref]
- Marsaud C, Rossignol S, Tounian P, Netchine I, Dubern B (2015) Prevalence and management of gastrointestinal manifestations in Silver-Russell syndrome. Arch Dis Child 100: 353–358. [Crossref]
- Yamaguchi KT Jr, Salem JB, Myung KS, Romero AN Jr, Skaggs DL (2015) Spinal deformity in Russell-Silver syndrome. Spine Deform 3: 95–97. [Crossref]
- Bruce S, Hannula-Jouppi K, Peltonen J, Kere J, Lipsanen-Nyman M (2009) Clinically distinct epigenetic subgroups in Silver-Russell syndrome: the degree of H19 hypomethyulation associates with phenotype severity and genital and skeletal anomalies. J Clin Endocrinol Metab 94: 579–587. [Crossref]
- Abraham MB, Carpenter K, Baynam GS, Mackay DJ, Price G, Choong CS (2015) Report and review of described associations of Mayer-Rokitansky-Küster-Hauser syndrome and Silver-Russell syndrome. J Paediatr Child Health 51: 555–560. [Crossref]
- Ghanim M, Rossignol S, Delobel B, Irving M, Miller O, et al. (2013) Possible association between complex congenital heart defects and 11p15 hypomethylation in three patients with severe Silver-Russell syndrome. Am J Med Genet A 161A: 572–577. [Crossref]
- Reik W, Walter J (2001) Genomic imprinting: parental influence on the genome. Nat Rev Genet 2: 21–32. [Crossref]
- Azzi S, Abi Habib W, Netchine I (2014) Beckwith-Wiedemann and Russell-Silver Syndromes: from new molecular insights to the comprehension of imprinting regulation. Curr Opin Endocrinol Diabetes Obes 21: 30–38. [Crossref]
- Eggermann T, Perezde Nanclares G, Maher ER, Temple IK, Tümer Z, et al. (2015) Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin Epigenetics 7: 123. [Crossref]
- Mackay DJ, Eggermann T, Buiting K, Garin I, Netchine I, et al. (2015) Multilocus methylation defects in imprinting disorders. Biomol Concepts 6: 47–57. [Crossref]
- DeChiara TM, Efstratiadis A, Robertsen EJ (1990) A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345: 78–80. [Crossref]
- DeChiara TM, Robertson EJ, Efstratiadis A (1991) Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64: 849–859. [Crossref]
- Begemann M, Spengler S, Gogiel M, Grasshoff U, Bonin M, et al. (2012) Clinical significance of copy number variations in the 11p15.5 imprinting control regions: new cases and review of the literature. J Med Genet 49: 547–553. [Crossref]
- Gronskov K, Poole RL, Hahnemann JM, Thomson J, Tümer Z, et al. (2011) Deletions and rearrangements of the H19/IGF2 enhancer region in patients with Silver-Russell syndrome and growth retardation. J Med Genet 48: 308–311. [Crossref]
- Brioude F, Oliver-Petit I, Blaise A, Praz F, Rossignol S, et al. (2013) CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J Med Genet 50: 823–830. [Crossref]
- Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, et al. (2015) Paternally inherited IGF2 mutation and & growth restriction. N Engl J Med 373: 349–356. [Crossref]
- Azzi S, Salem J, Thibaud N, Chantot-Bastaraud S, Lieber E, et al. (2015) A prospective study validating a clinical & scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet 52: 446–453. [Crossref]
- Zeschnigk M, Albrecht B, Buiting K, Kanber D, Eggermann T, et al. (2008) IGF2/H19 hypomethylation in Silver-Russell syndrome and isolated hemi-hypoplasia. Eur J Hum Genet 16: 328–334. [Crossref]
- Binder G, Seidel A-K, Martin DD, Schweizer R, Schwarze P, et al. (2008) The endocrine phenotype in Silver-Russell syndrome is defined by the underlying epigenetic alteration. J Clin Endocrinol Metab 93: 1402–1407. [Crossref]
- Eggermann T, Cappa M, Maghnie M, Loche S, Bottazzo GF (2009) Endocrine Involvement in Developmental Syndromes. Endocr Dev Basel 14: 10–19.
- Bruce S, Hannula-Jouppi K, Peltonen J, Kere J, Lipsanen-Nyman M (2009) Clinically distinct epigenetic subgroups in Silver-Russell syndrome: the degree of H19 hypomethyulation associates with phenotype severity and genital and skeletal anomalies. J Clin Endocrinol Metab 94: 579–587. [Crossref]
- Hall JG (2010) Review and hypothesis: syndromes with severe intrauterine growth restriction and very short stature—are they related to the epigenetic mechanism(s) of fetal survival involved in the developmental origins of adult health and disease? Am J Med Genet A 152A: 512–527. [Crossref]
- Akawi NA, Ali BR, Hamamy H, Al-Hadidy A, Al-Gazali L (2011) Is autosomal recessive Silver–Russel syndrome a separate entity or is it part of the 3-M syndrome spectrum? Am J Med Genet A 155A: 1236–1245. [Crossref]
- Binder G, Seidel AK, Martin DD, Schweizer R, Schwarze CP, et al. (2008) The endocrine phenotype in Silver– Russell syndrome is defined by the underlying epigenetic alteration. J Clin Endocrinol Metab 93: 1402–1407. [Crossref]
- Vu-Hong TA (2009) Russell–Silver syndrome with 11p15 epimutation: analysis of growth, bone maturation, puberty and response to GH treatment on a large series of 101 patients. Horm Res 72: 447-448.
- Epifanio M, Marostica PC, Mattiello R, Feix L, Nejedlo R, et al. (2012) A randomized, double-blind, placebo-controlled trial of cyproheptadine for appetite stimulation in cystic fibrosis. J Pediatr (Rio J) 88: 155–160. [Crossref]
- Chinuck R, Dewar J, Baldwin DR, Hendron E (2014) Appetite stimulants for people with cystic fibrosis. Cochrane Database Syst Rev 7: CD008190. [Crossref]
- Davies PS, Valley R, Preece MA (1988) Adolescent growth and pubertal progression in the Silver-Russell syndrome. Arch Dis Child 63: 130-135. [Crossref]
- Clayton PE, Cianfarani S, Czernichow P, Johannsson G, Rapaport R, Rogol A (2007) Management of the child born small for gestational age through to adulthood: a consensus statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J Clin Endocrinol Metab 92: 804–810. [Crossref]
- Ranke MB, Lindberg A (2010) Height at start, first-year growth response and cause of shortness at birth are major determinants of adult height outcomes of short children born small for gestational age and Silver– Russell syndrome treated with growth hormone: analysis of data from KIGS. Horm Res Paediatr 74: 259–266. [Crossref]
- Chernausek SD, Breen TJ, Frank GR (1996) Linear growth in response to growth hormone treatment in children with short stature associated with intrauterine growth retardation: the National Cooperative Growth Study experience. J Pediatr 128: S22–S27. [Crossref]
- Albertsson-Wikland, K (1989) Growth hormone secretion and growth hormone treatment in children with intrauterine growth retardation. Swedish Paediatric Study Group Growth Hormone Treatment. Acta Paediatr Scand Suppl 349: 35–41. [Crossref]
- Jensen RB, Thankamony A, O'Connell SM, Kirk J, Donaldson M, et al. (2014) A randomised controlled trial evaluating IGF1 titration in contrast to current GH dosing strategies in children born small for gestational age: the North European Small-for-Gestational-Age Study. Eur J Endocrinol 171: 509–518. [Crossref]
- Albertsson-Wikland K, Karlberg J (1997) Postnatal growth of children born small for gestational age. Acta Paediatr Suppl 423: 193–195.
- Smeets CC, Zandwijken GR, Renes JS, Hokken-Koelega AC (2016) Long-term results of GH treatment in Silver–Russell syndrome (SRS): do they benefit the same as non-SRS short-SGA? J Clin Endocrinol Metab 101: 2105–2112. [Crossref]
- Toumba M, Albanese A, Azcona C, Stanhope R (2010) Effect of long-term growth hormone treatment on final height of children with Russell–Silver syndrome. Horm Res Paediatr 74: 212–217. [Crossref]
- Rakover Y, Dietsch S, Ambler GR, Chock C, Thomsett M, et al. (1996) Growth hormone therapy in Silver Russell syndrome: 5 years experience of the Australian and New Zealand Growth database (OZGROW). Eur J Pediatr 155: 851–857. [Crossref]
- Willemsen RH, Arends NJ, Bakker-van Waarde WM, Jansen M, van Mil EG, et al. (2007) Long-term effects of growth hormone (GH) treatment on body composition and bone mineral density in short children born small-for- gestational-age: six-year follow-up of a randomized controlled GH trial. Clin Endocrinol (Oxf) 67: 485–492. [Crossref]
- Schweizer R, Martin DD, Schonau E, Ranke MB (2008) Muscle function improves during growth hormone therapy in short children born small for gestational age: results of a peripheral quantitative computed tomography study on body composition. J Clin Endocrinol Metab 93: 2978–2983. [Crossref]
- Ackland FM, Stanhope R, Eyre C, Hamill G, Jones J, et al. (1988) Physiological growth hormone secretion in children with short stature and intra-uterine growth retardation. Horm Res 30: 241–245. [Crossref]
- Tanner JM, Lejarraga H, Cameron N (1975) The natural history of the Silver–Russell syndrome: a longitudinal study of thirty-nine cases. Pediatr Res 9: 611–623. [Crossref]
- Latronico AC, Brito VN, Carel JC (2016) Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol 4: 265–274. [Crossref]
- Boonstra V, van Pareren Y, Mulder P, Hokken-Koelega A (2003) Puberty in growth hormone-treated children born small for gestational age (SGA). J Clin Endocrinol Metab 88: 5753–5758. [Crossref]
- Verkauskiene R, Petraitiene I, Albertsson Wikland K (2013) Puberty in children born small for gestational age. Horm Res Paediatr 80: 69–77. [Crossref]
- Leunissen RW, Oosterbeek P, Hol LK, Hellingman AA, Stijnen T, et al. (2008) Fat mass accumulation during childhood determines insulin sensitivity in early adulthood. J Clin Endocrinol Metab 93: 445–451. [Crossref]
- Leunissen RW, Kerkhof GF, Stijnen T, Hokken- Koelega A (2009) Timing and tempo of first-year rapid growth in relation to cardiovascular and metabolic risk profile in early adulthood. JAMA 301: 2234–2242. [Crossref]
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ (1989) Weight in infancy and death from ischaemic heart disease. Lancet 2: 577–580. [Crossref]
- Rich-Edwards JW, Stampfer MJ, Manson JE, Rosner B, Hankinson SE, et al. (1997) Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ 315: 396–400. [Crossref]
- Martyn CN, Barker DJ, Osmond C (1996) Mothers’ pelvic size, fetal growth, and death from stroke and coronary heart disease in men in the UK. Lancet 348: 1264–1268. [Crossref]
- Takenouchi T, Awazu M, Eggermann T, Kosaki K (2015) Adult phenotype of Russell–Silver syndrome: a molecular support for Barker–Brenner’s theory. Congenit Anom (Kyoto) 55: 167–169. [Crossref]
- Ong KK, Loos RJ (2006) Rapid infancy weight gain and subsequent obesity: systematic reviews and hopeful suggestions. Acta Paediatr 95: 904–908. [Crossref]
- Leunissen RW, Kerkhof GF, Stijnen T, Hokken-Koelega AC (2012) Effect of birth size and catch-up growth on adult blood pressure and carotid intima-media thickness. Horm Res Paediatr 77: 394–401. [Crossref]
- Sas T, Mulder P, Hokken-Koelega A (2000) Body composition, blood pressure, and lipid metabolism before and during long-term growth hormone (GH) treatment in children with short stature born small for gestational age either with or without GH deficiency. J Clin Endocrinol Metab 85: 3786–3792. [Crossref]
- van der Steen M, Lem AJ, van der Kaay DC, Bakker-van Waarde WM, van der Hulst FJ, et al. (2015) Metabolic health in short children born small for gestational age treated with growth hormone and gonadotropin-releasing hormone analog: results of a randomized, dose-response trial. J Clin Endocrinol Metab 100: 3725–3734. [Crossref]
- Van Pareren Y, Mulder P, Houdijk M, Jansen M, Reeser M, Hokken-Koelega A (2003) Effect of discontinuation of growth hormone treatment on risk factors for cardiovascular disease in adolescents born small for gestational age. J Clin Endocrinol Metab 88: 347–353. [Crossref]
- Feuk L, Kalervo A, Lipsanen-Nyman M, Skaug J, Nakabayashi K, et al. (2006) Absence of a paternally inherited FOXP2 gene in developmental verbal dyspraxia. Am J Hum Genet 79: 965–972. [Crossref]
- Augustine EF, Blackburn J, Pellegrino JE, Miller R, Mink J (2013) W. Myoclonus-dystonia syndrome associated with Russell Silver syndrome. Mov Disord 28: 841–842. [Crossref]
- Sheridan MB, Bytyci Telegrafi A, Stinnett V, Umeh CC, Mari Z, et al. (2013) Myoclonus-dystonia and Silver–Russell syndrome resulting from maternal uniparental disomy of chromosome 7. Clin Genet 84: 368–372. [Crossref]
- Kotilainen J, Hölttä P, Mikkonen T, Arte S, Sipilä I, Pirinen S (1995) Craniofacial and dental characteristics of Silver–Russell syndrome. Am J Med Genet 56: 229–236. [Crossref]
- Bergman A, Kjellberg H, Dahlgren J (2003) Craniofacial morphology and dental age in children with Silver– Russell syndrome. Orthod Craniofac Res 6: 54–62. [Crossref]
- Cullen CL, Wesley RK (1987) Russell–Silver syndrome: microdontia and other pertinent oral findings. ASDC J Dent Child 54: 201–204. [Crossref]
- Hodge N, Evans CA, Simmons KE, Fadavi S, Viana G (2015) Occlusal characteristics of individuals with growth hormone deficiency, idiopathic short stature, and Russell–Silver syndrome. J Dent Child (Chic) 82: 135–140. [Crossref]
- Giabicani E, Boule M, Galliani E, Netchine I (2015) Sleep apneas in Silver Russell syndrome: a constant finding. Horm Res Paediatr 84: 262.
- Bellver-Pradas J, Cervera-Sánchez J, Boldó-Roda A, Martín-Cortés A, Ferreres-Gómez L, et al. (2001) Silver–Russell syndrome associated to Mayer–Rokitansky–Kuster–Hauser syndrome, diabetes and hirsutism. Arch Gynecol Obstet 265: 155–157. [Crossref]
- Demars J, Gicquel C (2012) Epigenetic and genetic disturbance of the imprinted 11p15 region in Beckwith–Wiedemann and Silver–Russell syndromes. Clin Genet 81: 350–361. [Crossref]
- Begemann M, Spengler S, Gogiel M, Grasshoff U, Bonin M, et al. (2012) Clinical significance of copy number variations in the 11p15.5 imprinting control regions: new cases and review of the literature. J Med Genet 49: 547–553. [Crossref]
- Brioude F, Oliver-Petit I, Blaise A, Praz F, Rossignol S, et al. (2013) CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J Med Genet 50: 823–830. [Crossref]
- Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, et al. (2015) Paternally inherited IGF2 mutation and growth restriction. N Engl J Med 373: 349–356. [Crossref]
- Hoffmann K, Heller R (2011) Uniparental disomies 7 and 14. Best Pract Res Clin Endocrinol Metab 25: 77–100. [Crossref]
- Amor DJ, Halliday J (2008) A review of known imprinting syndromes and their association with assisted reproduction technologies. Hum Reprod 23: 2826–2834. [Crossref]
- Bowdin S, Allen C, Kirby G, Brueton L, Afnan M, et al. (2007) A survey of assisted reproductive technology births and imprinting disorders. Hum Reprod 22: 3237-3240. [Crossref]
- Eggermann T, Brioude F, Russo S, Lombardi MP, Bliek J, et al. (2015) Prenatal molecular testing for & Beckwith – Wiedemann and Silver – Russell syndromes: a challenge for molecular analysis and genetic counseling. Eur J Hum Genet 24: 784-793. [Crossref]
- Giabicani E, Netchine I, Brioude F (2016) New clinical and molecular insights into Silver-Russell syndrome. Curr Opin Pediatr 28: 529-535. [Crossref]