Pathogens of Emerging Infectious Diseases (EIDs) have sometimes caused serious public health threats. Researchers have put in significant efforts to not only explore and improve the existing therapeutic methods but also prevent the spread of the pathogen. The present COVID-19 pandemic requires administrators and healthcare professionals to make unprecedentedly difficult and rapid countermeasures. The most difficult challenge is that asymptomatic people with infection can sometimes cause clusters of infections. Identification and tracking have always been delayed since diagnostic confirmation relies solely on real-time reverse transcription polymerase chain reaction (RT-PCR) analysis, which requires a long time for the analysis and sometimes gives false positive or negative results, creating further confusion to administrators and healthcare professionals and increasing the risk of infection. This fact indicates that one of the most important keys of infection control when encountering a new EID, including the latest COVID-19, is the extensive use of an effective diagnostic device that can rapidly characterize the pathogen and provide quantitative information. It should be noted that serious respiratory infections, such as COVID-19 and SARS, and also some foodborne, zoonotic, animal, and plant infectious diseases, are a serious threat to the general public. In particular, mutations in zoonotic pathogens that cause human diseases such as H5N1 and H1N1 would be a greater threat to the human society both now and in the future. Countermeasures against these infectious diseases will strongly rely on rapid and precise diagnosis, and the development of diagnostic devices is very crucial. This review describes the requirements and the issues pertaining to diagnostic devices, strategies to achieve ultra-high sensistivity, and the principles of the recently developed analytic methods, for the benefit of frontline users, including healthcare professionals and the representatives of the diagnostic equipment at the institutions.
EID, SAR, COVID, Ebola
Pathogens of Emerging Infectious Diseases (EIDs) have sometimes caused serious public health threats, such as not only the historical Spanish flu, but also the recent severe acute respiratory syndrome (SARS) and the Ebola outbreaks. Researchers have put in significant efforts to not only explore and improve the existing therapeutic methods but also prevent the spread of the pathogen [1]. However, the present COVID-19 pandemic requires administrators and healthcare professionals to make unprecedentedly difficult and rapid countermeasures.
The most difficult challenge is that asymptomatic people with infection can sometimes cause clusters of infections. Identification and tracking have always been delayed since diagnostic confirmation relies solely on polymerase chain reaction (PCR) analysis, the number of devices and operators, which have been insufficient. Real-time RT-PCR requires a long time for the analysis and sometimes gives false positive or negative results [2-9], creating further confusion to administrators and healthcare professionals and increasing the risk of infection.
This fact indicates that one of the most important keys of infection control when encountering a new EID, including the latest COVID-19, is the extensive use of an effective diagnostic device that can rapidly characterize the pathogen and provide quantitative information. The challenge in the diagnostic confirmation of EIDs is mainly due to two reasons. The genomic information of the pathogen is unknown and the antibody is, in fact, not prepared, both of which seriously undermine the control interventions to prevent spread at the initial stage of outbreak. The former and the latter are related to PCR and immune reaction-based assays, respectively. In particular, the antibody is essential to prepare the virus detection test kit based on immunochromatography, which is indispensable for rapid inspection at the frontline of infection defense.
It should be noted that serious respiratory infections, such as COVID-19 and SARS, and also some foodborne, zoonotic, animal, and plant infectious diseases, are a serious threat to the general public. For example, norovirus and the pathogenic Escherichia coli have often caused outbreaks in local communities [10]; avian flu, swine flu, and African swine fever have sometimes devastated the poultry and pork industries in several countries [11-14] In particular, mutations in zoonotic pathogens that cause human diseases such as H5N1 and H1N1 would be a greater threat to the human society both now and in the future [15-17].
As described previously, countermeasures against these infectious diseases will strongly rely on rapid and precise diagnosis. Therefore, the development of diagnostic devices is very crucial. This review describes the requirements and the issues pertaining to diagnostic devices, strategies to achieve ultra-high sensitivity, and the principles of the recently developed analytic methods, for the benefit of frontline users, including healthcare professionals and the representatives of the diagnostic equipment at the institutions.
Social needs and requirements for diagnostic devices
Social needs and general requirements: Here, we consider the social needs and requirements for the diagnostic devices to develop countermeasures against the newly emerging pathogens. The diagnostic devices for pathogens are used by a wide variety of institutions, including hospitals, health care centers, private diagnostic companies, food plants, food distributors, ranches, and farms. As mentioned above, not only human infectious diseases but also zoonotic, animal, and plant infectious diseases have sometimes caused significant social and economic losses. Even if the same kind of diagnostic device is used, the operations during normal times and when facing the outbreaks should be different.
During normal times, it is mainly used for the detection and surveillance of pathogens, which is important for the appropriate selection of antibiotics or antiviral agents for patients and for the prevention of the outbreak. Therefore, the operations can be carried out by experts at the usual place with minimal difficulties. In contrast, during an emergency, there are many challenges faced by institutions. Firstly, a large number of the samples should be inspected with the highest urgency and hence, sufficient number of diagnostic devices should be procured, even though they require the individual reagent kits and protocols. Secondly, the operation of the devices will sometimes be carried out by non-experts in temporary spaces in poor environments, making the process even more difficult.
These social needs and operational difficulties reveal the general and technical requirements for diagnostic devices. The devices are often manufactured under the pressure of stiff sales competition and hence, the commercially available devices sometimes require analysis protocols and/or reagent kits specific to them, even based on the same principle. These situations are unfavorable during an emergency to make countermeasures against very rapid expansion of the infection, so that the specific device, even if it displays very high performance, might be not helpful. Accordingly, manufacturers are required to provide, at least, the pretreatment and reaction kits, and the quantification software, with sufficient commonality to fulfill their social responsibility.
One should remember that the success of the diagnosis must be determined not by the device itself and its principle, but by the properties of the primers or antibodies specific for the target pathogen. Therefore, the research and development activities of the suitable primers and antibodies should be very significant and prepared for the possible mutations of the pathogens that might occur [18-21].
Technical requirements: The analytical principles of the pathogens can be roughly divided into two types of measurements. One is based on the genetic information, that is, DNA or RNA, of the target pathogen, and the other is based on the antibodies that specifically recognize the target pathogen, that is, the ligand binding assay (LBA). Based on the two analytic principles and the social and general requirements described above, we consider the technical requirements for the diagnostic devices.
In general, assays of the samples from patients and individuals suspected of being infected are much more difficult than those for research purposes since they usually contain a very small number of the target pathogen and/or the key related compounds such as the DNAs or RNAs and the antibodies, with a very large amount and variety of contaminants, consisting of proteins, lipids, carbohydrates, and electrolytes in addition to the DNA, living, and dead bacteria unrelated to the infection. Moreover, even though the collection procedure of the sample is ensured, the compositions of the individual samples, which should be collected by different staff, may be subtly different from each other [22].
In the case of DNA/RNA-based assays, the DNAs and RNAs unrelated to the target pathogen may cause an increase in the background signal and turn out to be false-positive or false-negative, at worst [23]. In the case of antibody-based assays, the contaminants may cause the nonspecific binding to increase in the background signal, thereby reducing the confidence of the assay [24-26]. Therefore, even if a superior diagnostic device is developed, the pretreatment protocol of the sample may be a bottleneck for the assay. This is the reason why most of the efforts have been expended to improve the pretreatment protocol by the manufacturers as the key step of the development. In contrast, academic researchers, including us, have been developing a novel principle and methodology of the assay with scientific interest. The innovative new principle should be the key technique to realize next-generation diagnostic devices.
So far, we have described the current scenario of the pathogen assays to reveal the subjects to be addressed. In the next section, we describe the features of the recently developed high-performance diagnostic methods, including our recent results. Some of them have been commercially available and used as the key tool for preventing the spread of COVID-19.
2. Strategies to achieve high-performance of pathogen assays
It is clear that the conventional real-time PCR systems, which are currently used as the main diagnostic device to fight the COVID-19 pandemic, are not sufficient to address the general and technical requirements described above. A much higher sensitivity, throughput, and reliability need to be achieved [27]. It should be noted that digital detection has made breakthroughs in assay methodologies, which are closely correlated with the following key strategies. In the DNA/RNA-based assays, digital PCR [28-31] and Laser PCR® [32] have made significant leaps in throughput. In the ligand binding assay (LBA), the digital detection of the ligand-analyte (pathogen) interaction such as the ON or OFF signal enables the assay to allow for ultra-high sensitivity, effective elimination of the background signal, simultaneous multiplatform, and ultra-high throughput. A typical outcome is the digital ELISA based on the module for imaging single molecule arrays ‘SiMoA’ [33-35]. Digital detection systems require highly homogeneous discrete microenvironments such as reaction and/or detection platforms, such as microdroplets, microbeads, nanoparticles, microwells, and microarrays, which are sometimes combined and used together. Practical examples based on digital detection, including our recent results, are described in the latter sections.
The improvement of the throughput can be realized by diminishing the sample volume. This strategy is particularly effective in reducing the PCR cycle time by decreasing the heat capacitance of the sample and the device. Moreover, diminution of the sample volume leads to more rapid and noninvasive sampling from patients and individuals suspected of being infected, and is also cost-effective as it requires only lesser amounts of the reagent. Simultaneously, however, the signal intensity should be remarkably reduced, and the noise and background signals may disturb the detection correspondingly. The signal-to-background (S/B) ratio is sometimes more important than the signal-to-noise (S/N) ratio in the assay of a very small amount of the analyte. Accordingly, the practical detection limit depends not only on the photodetector itself, which has been remarkably improved and single-molecule level detection can be easily performed in recent diagnostic devices [36], but also on the effective reduction of the optical background signal.
One of the most effective methods for reducing the optical background is disuse of the excitation light. The chemiluminescence (CL) or bioluminescence (BL)-based assays are typical examples, while additional procedures, such as the addition of the luminescence substrate and the enhancer, are required in the assay, and the resulting luminescence intensity may be strongly time-dependent, preventing precise quantification. Electrochemiluminasence (ECL) is an alternative method to the conventional CL or BL [37]. The practical assay system, that is, the electrochemiluminescence immunoassay (ECLIA), has been employed as a powerful diagnostic device for the quantification of the COVID-19 antibodies [38].
In next-generation diagnostic devices, photoexcitation will still be very effective not only for detection but also for the heating of the polymerase, such as photo- or laser-PCR. From this point of view, the application of near-field optical techniques has remarkably contributed to the development of next-generation diagnostic devices, in which the electronic excitation or heating at the nanospace allows for the realization of ultra-fast PCR and ultra-high sensitivity immunoassay. Here, we will describe the principle and features of next-generation diagnostic devices (Scheme 1).
Assay based on quantification of the pathogen gene
Improvement of PCR system based on photothermal cycler: PCR analysis involves a large number of thermal cycles, consisting of three steps, namely denaturation, annealing, and extension, conducted at different temperatures and times [39]. Each cycle requires a rather long time to achieve satisfactory thermal equilibrium and reaction yields; therefore, the time for the assay to be performed is at least 20-30 min. As mentioned above, the diminution of the sample volume is an effective strategy to achieve high throughput [40]. In practice, there are two practical approaches. One is the application of microfluidic devices combining microvolume reactors, and the other is the division of the sample to microdroplets, both of which is based on photothermal cycler, and can be combined with digital PCR. A more advanced and effective heating method is plasmon heating at the nanospace, which is one of the hottest research topics in near-field optics, and the practical examples will be described later.
PCR based on infrared (IR) heating: The idea of IR heating to drive PCR has been reported many years ago using a conventional tungsten lamp [41-44]. In this method, thermal cycling in the microfluidic system is usually carried out on the stage of the microscope by using IR light through an IR long-pass filter (Figure 1), so that the simultaneous fluorescence measurement for the confirmation and/or quantification of the PCR is successfully carried out during the thermal cycling (Figure 2). It is to be noted that for parallel heating of several PCR vessels in the microfluidic device, extremely homogeneous illumination covering a wide area in the microchip without any interference fringes or shadows is required. An incoherent light source such as a classical tungsten lamp or a near-IR LED is more effective than the near-IR laser for this kind of application. The demonstration of the simple PCR in the microfluidic chip in the initial studies on this methodology, as well as that of the quantitative PCR and practical diagnostic assays of the influenza virus based on RT-PCR, have been reported [43,44]. The virus assay of a mock nasal swab sample at clinically relevant concentrations was carried out using a carefully designed, integrated two-chip system consisting of solid-phase extraction (SPE) and RT-PCR systems [43]. As mentioned above, the pretreatment of the sample is often one of the most significant factors to achieve precise diagnosis by the effective selection of the pathogen from the contaminants. In the stand point of view, such a combination of pretreatment and analysis units may be a good solution to ensure flexibility in the design of the assay system to confront many kinds of pathogens, for example, viruses and bacteria. A similar IR-induced quantitative PCR system has also been reported by other groups [45-48], in which they have expanded the applicability of the IR-PCR to quantitative determination of the initial copy number by integrating fluorescence detection during the amplification process [45] (Figures 1 and 2).
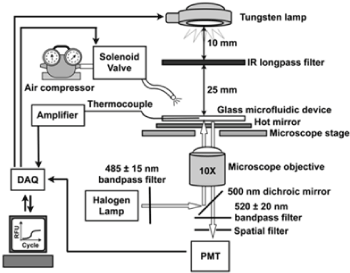
Figure 1. Layout of the infrared heating and fluorescence detection systems. The temperature control system consisted of a W-lamp as the IR source, a thermocouple for temperature measurement, a compressed air source for cooling, and electrical relays used to turn on and off each of these components. The fluorescence system used a halogen lamp for excitation and a photomultiplier tube (PMT) for detection. A 775 nm long-pass IR filter and hot mirror were used to decouple the two optical systems. Reproduced from [47] with permission from The American Chemical Society.
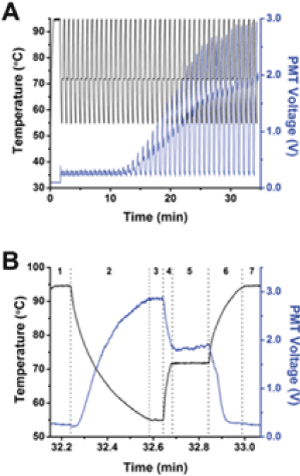
Figure 2. Simultaneous collection of temperature and fluorescence. In all graphs, temperature is plotted as the black line and corresponds to the left y-axis, and PMT voltage from SYBR Green fluorescence is plotted as the blue line and corresponds to the right y-axis. (A) 107 starting copies of PUC19 were amplified by PCR in 40 cycles. The fluorescence intensity varied as a function of temperature and cycle, but the overall trend indicated that the intensity increased starting at approximately the 14th cycle and reached a plateau at approximately cycle 30. (B) A single temperature cycle and fluorescence data from part A are shown. As explained further in the text, the stages are shown at the top of the figure and correspond to different times during the thermal cycling, such as denaturation, annealing, or extension. Reproduced from [47] with permission from The American Chemical Society.
3.1.2 Laser heating PCR in microdroplets on a Petri dish
More effective laser heating PCRs in microdroplets have been demonstrated by some groups. For example, the illumination of a 1.46 μm diode laser light to the nanoliter droplets arrayed on a Petri dish [49]. A large array of uniform-sized nanoliter droplets of the PCR mixture with a diameter of 100 μm at pitches of 250 μm or less between droplets were generated on a polystyrene Petri dish by a simple contact printing technique under careful humidity control. After printing, the array was permanently protected from evaporation by adding mineral oil around the array until the array was covered (Figure 3, Figure 4a). The use of this mineral oil, which does not absorb 1.46 μm light, should be effective for rapid photoheating and rapid annealing of the microdroplets upon photo-thermal cycling. The beam size was measured to be ~ 200 μm at FWHM with a 10X objective lens. The temperature of the droplets was controlled by the laser power, and the dependence was calibrated using a fluorescence probe and the yields of the PCR amplicons. For a typical droplet with a diameter of ~300 μm, 25 and 50 mW of laser power were required for the annealing/extension and melting steps, respectively. The optimized conditions were a combination of an initial 10 s for enzyme activation, 2 s for melting, and 8 s for annealing/extension, respectively, allowing completion of 40 amplification cycles in ~6 min. Moreover, clear time-dependent amplification curves were obtained from the series of different template DNA concentrations, which can be used for real-time PCR as well as sufficiently intense end-point fluorescence intensities than the baseline (Figure 4). Although this work seems very attractive for many critical applications such as the amplification of small amounts of template DNA and single cell assays, the most important and attractive aspect should be the application to digital PCR, which requires the division of the sample solution into a large number of highly homogeneous discrete microdroplets before the PCR cycles, as described later (Figures 3 and 4).
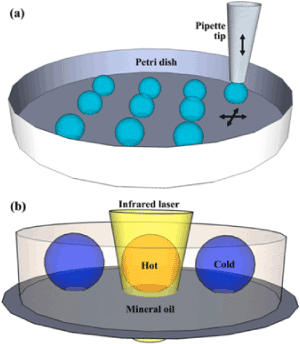
Figure 3. A IR laser induced PCR on a Petri dish; (a) Illustration of the contact printing on a Petri dish. A disposable pipette tip is vertically controlled to make a brief contact with the substrate leaving a droplet on the surface. The Petri dish is positioned by the dual-axis motorized linear stages. (b) Illustration of the infrared laser heating of a droplet in the oil phase using an inverted microscope. Reproduced from [49] with permission from The Royal Society of Chemistry.

Figure 4. Bright field (a) and fluorescence (b) images of a 4 by 5 array of PCR mixture droplets. 10 droplets in the upper right were PCR amplified while the rest were not amplified. Reproduced from [49] with permission from The Royal Society of Chemistry.
Digital PCR
Real-time PCR has been widely used for gene expression analyses in biological studies. The gene copy number contained in the sample can be accurately determined by comparing the position at the beginning of the exponential phase, the so-called cycle threshold (Ct), with a series of standard samples containing the target genes [50,51]. In diagnostic use, the gene numbers of the target pathogen in the samples from patients and individuals suspected of being infected can be directly determined in the absence of any immunological information of the pathogen. Therefore, real-time RT-PCR has been used as a main tool for the diagnosis of the present COVID-19 outbreaks and pandemic. Despite numerous advantages, however, conventional real-time PCR has some disadvantages in terms of quantification reliability and low throughput. Quantification depending on the standard samples is not suitable for the detection of ultra-small amounts of the target genes desired in the pathogen assay, and performing several PCR cycles may be time consuming.
Digital PCR is a long-awaited novel approach that can address problems related to conventional real-time PCR [28-31,52-54]. The most characteristic feature of digital PCR compared to conventional real-time PCR is the digital counting of the huge numbers of PCR results, that is, success (ON=1) or failure (OFF=0), in the discretely partitioned microsamples prepared from the same sample. During amplification, fluorescent dye-labeled probes were used to detect the target genes. When the microsample contains the target gene, even if it is a single molecule, the PCR to the endpoint level affords sufficient amount of the amplicon to give the intense fluorescence signal as “On”. When the microsample does not contain the target gene, on the other hand, no amplicon is produced, so that the fluorescence signal is not observed keeping the baseline level as “Off”. Following PCR analysis, the fraction of negative reactions was used to generate an absolute count of the number of target molecules in the sample, without the need for standards or endogenous controls. To account for wells that may have received more than one molecule of the target gene, a correction factor is applied using the Poisson model [31,55]. It should be noted that the most important factor of this assay is the suitable preparation of the microsamples to contain very small amounts of the target gene. In the theoretical calculation, the optimized number of genes in each partition should be 1.6 molecule. Within the pathogenic assays, however, the usual sample contains a very small amount of the target pathogenic genes and one may not pay much attention to this point (Figure 5).
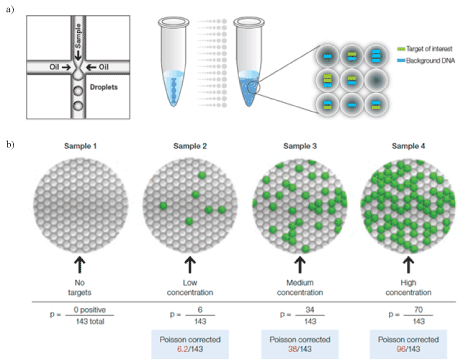
Figure 5. Principle of digital PCR [31]: a single PCR sample is partitioned into 20,000 discrete nanoliter-seized droplets by a droplet generator. Target and background DNA are distributed randomly into the droplets during the partitioning process, (a). Droplets are assigned as positive or negative by thresholding based on their fluorescence amplitude. The number of positive and negative droplets is used to calculate the concentration of the target and reference DNA sequences and their Poisson-based 95% confidence intervals, (b). Reproduced from [31] with permission from Bio-Rad Laboratories, Inc.
3. 3 Advanced PCR based on nanophotonics
The concept of digital PCR is the digital counting of PCR results from the large numbers of discretely partitioned microsamples, which are provided by a microdroplet generator or a microfluidic device having arrayed microvessels. Although the concept of digital PCR is highly advanced, most of the commercially available systems employ the old-fashioned conventional thermal cycler [50] instead of advanced photoheating. Therefore, thermal cycling may be a nasty bottleneck to improve the throughput, which is required as much as possible, particularly upon the outbreak and pandemic of EDIs. Fortunately, the remarkable advances in nanophotonics provide a very effective and revolutionary solution, that is, surface plasmon (SP) heating based on the photon-phonon conversion.
SP is the photoelectromagnetic field localized on the metal surface [56]. There are two types of SPs: one is the propagating SP on a flat metal surface, and the other is the localized SP (LSP) on the nanosized metal particles or nanostructures having sharp edges, gaps, pleats, and both SPs can be easily excited by visible and near-IR light. Since the SP fields are strongly localized on the metal surface and are effectively converted to heat energy, one can expect a very rapid and effective photon-electron-phonon conversion [57,58]. In other words, SPs can be used as nanosized heaters on the surface by which one can selectively heat molecules immobilized on the surface or very close to the surface. It is impossible to achieve such selective heating in the nano-sized area by conventional light illumination from the far field, even using a conventional microscope. Accordingly, SP heating should be matched very well with advanced PCR techniques requiring effective heating at the nanospace to realize ultra-high sensitivity and throughput. The two practical applications are described as follows.
Photonic PCR
Advanced PCR based on photonic heating has been reported [59]. Although the excitation of the SP on a flat metal surface usually requires a prism coupler and a p-polarized laser, it can also excite the SP by fine-tuning the thickness of the metal film. In this case, the characteristic plasmonic absorption band similar to that of the LSPs on the nanoparticles appears on the gold film, which can be excited by a conventional light source such as an LED without a prism coupler. For example, 65% of the excitation light at the peak wavelength (450 nm) of the excitation LEDs was absorbed in the 120 nm Au film. Based on this concept, a photonic PCR system with a simple optical configuration can be constructed on the PMMA substrate with PCR wells, in which the Au film is evaporated on the bottom surfaces, set on the excitation LED arrays. The typical PCR thermal cycling, consisting of three representative temperatures (94°C for denaturation, 60°C for annealing, and 72°C for extension), was performed 30 times in 5 min.
The amplification of l-DNA was performed to verify this photonic PCR method, and the 98-bp amplicon was selectively obtained by 30 and 60 cycles, similar to the PCR using a conventional thermal cycler, demonstrating good thermal accuracy of this photonic PCR. The yields of the amplicons were rather poor than those of the conventional PCR since the heat capacitance of the thin Au film may be too small to induce sufficient thermal diffusion into the bulk solution. This limitation can be overcome by two approaches: one is the utilization of a three-dimensional (3D) photonic substrate in the PCR chamber for uniform photothermal heating of the PCR mixture, and the other by the direct immobilization of the target DNA on the Au surface, as demonstrated in our approach described in section 3.1.1. The thermal response of the flat metal film with a rather large heat capacity is much slower than that of the nanoparticles in Laser-PCR® described in the following section, while the good uniformity of the illumination should be adapted to the microfluidic device to achieve high throughput. Moreover, SP on the flat metal surface is effective not only for heating but also for the fluorescence detection of the target molecules by the specific fluorescence enhancement effect and the suppression of the excitation light [60-62] (Figure 6).
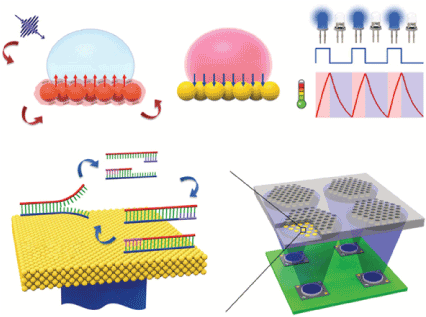
Figure 6. Ultrafast photonic PCR. (a) Schematic of the plasmonic photothermal light-to-heat conversion and subsequent heating of the surrounding solution (here, the PCR mixture) through ultrafast photon–electron–phonon couplings. When a light is turned off, fast cooling of heated solution can be achieved by the heat dissipation through the thin Au film. (b) Schematics of the ultrafast photonic PCR using a thin gold (Au) film as a light-to-heat converter and excitation light from the LEDs. Thermal cycling, consisting of two or three discrete temperatures for denaturation, annealing and extension, is required for nucleic acid amplification through the PCR. For multiple PCR reactions, each LED could be modulated separately so that there are unique annealing temperatures for each primer design. LED, light-emitting diode; PCR, polymerase chain reaction. Reproduced from [59] with permission from Springer Nature Limited.
Laser-PCR®, LSP-induced digital PCR: Nano-sized metal particles have been known to show characteristic color, and this fact has been known since the Middle Ages [63]; for example, the stained glasses in the cathedrals are typical applications of metal colloids. However, the reason for the coloring has been scientifically elucidated not too long ago. The deep-red color of gold colloids with a very large extinction coefficient is due to the plasmonic oscillation of the free electrons confined in the nanoparticles and coupled with the green light with a wavelength of approximately 520 nm, resulting in the formation of the LSP [64,65]. In other words, green light with a wavelength of 520 nm can be confined to nano-sized (approximately 5-20 nm) gold colloids. Since the resulting LSP is rapidly converted to phonons and the heat capacity of the nanoparticles is very small, the selective heating of the molecules immobilized on the surface or very close to the particle can be expected upon photoexcitation, whereas the temperature of the bulk solution surrounding the nanoparticles should be unchanged. This site-selective heating effect based on LSP, which is sometimes called a ‘nano-stove’, has been intensively studied in recent years and mainly applied during hyperthermia treatment of cancer cells using surface-modified gold particles with molecules recognizing cancer cells [57,66].
Laser-PCR® [32] is, however, a much more attractive and effective biomedical application of LSP because it can realize the next-generation of digital PCR. In Laser-PCR system, the micro-partitioning of the sample mixture for the digital PCR is conducted by the use of the nanosized gold particles, on which the primers for PCR are immobilized and act as individual ‘vessels’ for digital detection [32]. Suitable temperatures for the melting, annealing, and elongation of the DNAs can be readily obtained by controlling the laser power, so that sufficient PCR cycles can be completed in a very short time, that is, a million times faster than that required by a conventional thermal cycler. Moreover, the bulk solution around the particles is not heated at all, as mentioned above, which is particularly significant for digital PCR because a background level as low as possible is essential for digital detection of the PCR results [32]. Laser-PCR® with ultra-high throughput and wide applicability, therefore, should be one of the most expected diagnostic devices for countermeasures against the current and forthcoming EIDs (Figure 7).
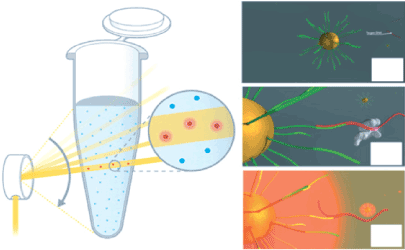
Figure 7. Schematic depiction of pulse controlled amplification of nucleic acids by Laser- PCR®; Laser-PCR® operates on the principle of pulse controlled amplification (PCA). A high-power laser beam is rapidly pulsed through the PCR reaction to selectively and specifically irradiate gold nanoparticles. Heat generated by the nanoparticles during laser pulsing dissipates directly into the solution by a steep temperature gradient, effectively heating up only primers or amplicons bound to their surface. As soon as the laser beam stops illuminating the nanoparticles, they immediately cool down to the set temperature of the reaction solution. Therefore, the bulk liquid of the reaction serves as a built-in cooling reservoir. Rendered images (Panels A, B and C) show the Pulse Controlled Amplification principle on the nano scale. Panel A) In the Laser-PCR®. reaction mix, gold nanoparticles (yellow) are functionalized with target-specific primers (light green); (B) the DNA target (red) anneals to the specific primer on the gold nanoparticle, and DNA polymerase binds for elongation; (C) a laser beam scanned over the sample excites all nanoparticles leading to localized heating and denaturation of amplicons. Effectively, this reaction follows a two-step temperature protocol, allowing nearly instantaneous thermal ramps and ultra-fast PCR reaction times. Reproduced from [32] with permission from De Gruyter.
The antibody-based assay
The development of a rapid detection chip for the currently expanding virus is particularly significant for preliminary diagnosis. Despite the important prerequisite that the highly specific antibody to the pathogen must be prepared, the antibody-based assay is one of the best options for preparing diagnostic devices for EIDs because of their high selectivity, sensitivity, and convenience. In this section, we describe the novel concepts and principles of advanced immunoassay systems.
Ultra-high sensitive ELISA: The development of ELISA has resulted in a revolution in bioassays [67]. The enzyme-catalyzed reaction amplifies the detection signal from the single antibody-antigen interaction by the turn-over of the enzyme cycles, which can be over tens of thousands depending on the reaction time. It is easy to see that, however, the throughput and sensitivity, which reaches up to 50 pM [68], are in tradeoff with each other. In other words, one can remarkably improve the throughput by improving the detection sensitivity. The sensitivity is remarkably improved by the use of highly sensitive optical effects, such as fluorescence, chemiluminescence, and bioluminescence, instead of the conventional colorimetric detection of the colored end product [69]. The features and limitations of these detections are outlined as follows.
It is well known that fluorescence detection generally provides a much higher (10–1000-fold) sensitivity than the conventional colorimetric detection, and the detection of even a single molecule is allowed without great difficulty. In the ELISA, the peroxidase produces several numbers of the fluorescent product by the enzyme cycles, so that the sensitivity is further enhanced compared to direct or indirect immunofluorescence [70,71]. One of the limitations of fluorescence detection is the poor quantification quality in the high concentration region, but this may not usually cause problems in the pathogen assay. Fluorescence detection requires intense excitation light, which causes photobleaching of the fluorescent products and background signal, and it is to be noted that both of them sometimes disturb the detection of a very small amount of the target antigen. The single-molecule counting technique (SMC®) [72] is an excellent method that addresses the disadvantages of fluorescence detection [73]. The characteristic feature of this method is the digital counting of the numbers of secondary fluorescent dye-labeled antibodies released into the elute from the sandwich immune complexes formed on the magnetic beads by chemical treatment [73]. Using this method, the presence of the secondary antibody in the elute can be detected as a flash signal of the fluorescence before photobleaching, and the background signal from the nonspecific interaction can be effectively eliminated. The employment of chemiluminescence and bioluminescence for detection also addresses such limitations of the fluorescence attributed to the excitation light [74]. The ELISAs based on chemiluminescence and bioluminescence were named CLEIA [71,75,76] and BLEIA [77-79], respectively. However, the advantage brings with it a few disadvantages; further steps for the induction of luminescence should be added to the general protocol of ELISA, which is already complex and takes a long time (Figure 8).
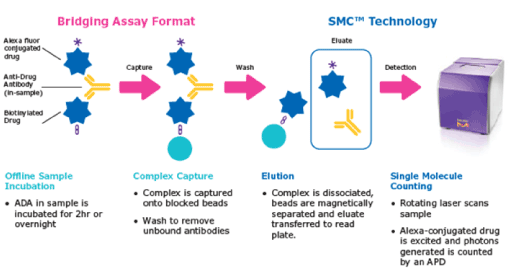
Figure 8. A SMCTM system detects fluorescence of the dye-labeled antibodies released from the immune complexes after a sandwich immunoassay. Digital detection of the single molecule fluorescence pulses effectively eliminates the background fluorescence under the detection threshold. Reproduced from [72] with permission from Merck KGaA.
Electrochemiluminescence immuno assay (ECLIA): The electrochemiluminescence immunoassay (ECLIA) is a relatively newly developed method that provides ultrasensitive detection of the analyte without the complex protocols required in ELISA [80-84]. In ECLIA, it is not the enzyme cycles but the electrochemical redox cycles that produce a large number of the luminescent species from the substrate. In the sandwich assay, the second antibody having a so called ‘sulfo-tag’ is employed to induce repetitive electrochemical oxidation of the Ru(II) complex in the solution, resulting in the Ru(III) complex. A sacrificial electron-donating substrate such as tripropylamine (TPA) in the solution is also oxidized to TPA+, giving the highly reductive radical species (TPA*) by rapid deprotonation. The recombination between the Ru(III) complex and TPA* generates the excited state of Ru(II) (Ru(II)*), exhibiting a characteristic intense orange luminescence. The use of TPA for the indirect recombination of the Ru complex is necessary to avoid the electrochemical splitting of water on the electrode in an aqueous medium, which is mostly used for analytical applications. By this cycle, the binding signal can be amplified and detected with a very high sensitivity in a wide concentration range of the target antigen. Moreover, the recombination resulting in luminescence occurs in the solution apart from the binding site of the sulfo-tag on the electrode, so that the background and the nonspecific signals can be effectively suppressed. This feature is particularly effective for the assays of the samples, including blood plasma serum without rigorous pretreatment. The assay kit for the detection of anti-SARS-CoV-2 by ECLIA has been commercially available from Roche [38].
Although the detection of DNA by ECLIA is possible and applied for some biological studies using a digoxigenin (DIG) - labeled probe DNA/RNA, the direct detection and quantification of the pathogenic genes has not been reported at present. The sensitivity may improve to achieve direct detection in the near future. The disadvantages of the ECLIA are mainly due to the necessity of the electrode prepared on the bottom surfaces of the wells, which makes the price of the multi-well plate more expensive than that of the conventional ELISA plate.
The microfluidic device integrating the ECLIA unit is particularly expected to realize digital detection transcending the properties of the digital ELISA, whereas the cost for the assay should be rather expensive because the production cost of the complex assay platform including microelectrodes will increase (Figure 9).
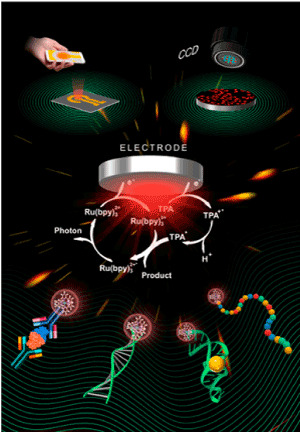
Figure 9. Principle of electrochemiluminescence based bioassays. Reproduced from [83] with permission from The American Chemical Society.
The digital ELISA: The digital PCR provides a much higher performance than conventional PCR, as described in the above section. In the immunoassay, the digital detection of the antigen-antibody interaction as the ON or OFF signal is also expected to improve the sensitivity and throughput, and to suppress the background signal and the consumption of expensive reagents simultaneously. The idea of digital detection has been presented about a decade ago and has been realized in practical diagnostic systems. The presented systems are principally based on the microbeads as the ‘solid phase’, on which the primary antibody is immobilized. After the reaction with the sample, the microbeads were treated with the fluorescent dye-labeled secondary antibody, washed, delivered into the discrete microcavities or microwells prepared in the microfluidic device, and analyzed by two-dimensional imaging of the fluorescence, giving the digital data. Although the principle is simple, there are many difficulties in each process to realize the practical level. Therefore, only a few systems are commercially available at present. One of the successful products is the single-molecule array [33-35], ‘SiMoA’, which is employed not only as the ultra-high sensitive ELISA getting closer to the attomolar sensitivity [85] but also for advanced clinical applications such as single-cell diagnosis of the diseases [86] (Figure 10).
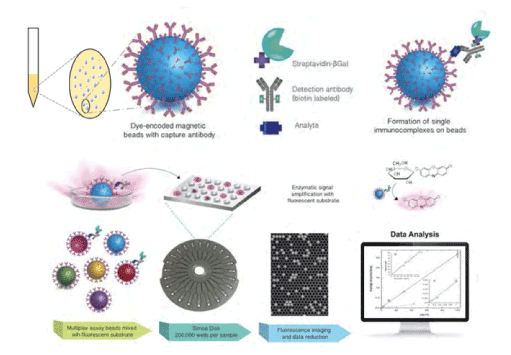
Figure 10. Ultra-sensitivity through ‘Simoa®’, Courtesy of Quanterix Corporation. Principle of ‘SiMoA®’ digital ELISA system. Paramagnetic SiMoA® beads are used as the solid phase in a sandwich immunoassay containing an enzymatic label. The labeled beads are mixed with a fluorogenic substrate and loaded into an array of femtoliter-size wells on the SiMoA® disc, where the beads settle into individual wells. The wells are sealed with a layer of oil, which prevents the beads from diffusing out of the wells and allows the fluorescent enzyme product to accumulate at high concentrations, enabling detection of a single enzyme molecule. Fluorescence imaging and data reduction is used to calculate the concentration of each analyte in the assay. This figure and caption are Courtesy of Quanterix® Corporation.
Ultra-sensitive detection of the pathogen assisted by SP enhanced isothermal DNA elongation: As mentioned in the laser-PCR section, SP is very effective for site-selective heating at the scale of single particles or single molecules. We have presented a novel method of digital detection of antigen-antibody complexes wrapped with DNA, which are isothermally elongated by the ‘molecular-selective’ heating based on the surface plasmon resonance (SPR) phenomenon [87]. The most characteristic feature of this method is the employment of isothermal rolling circle amplification (RCA) of the DNA, which is totally unrelated to the pathogen gene itself, for the selective signal amplification of the pathogen-antibody binding on the substrate. In RCA, the replication isothermally occurs from a small primer without any temperature change by using a circular template and a polymerase with strand displacement activity.
The concept of this method is described as follows, with the experimental procedures in some detail to be easily understandable. The plasmonic medium, a 50 nm Au film, was prepared on the glass substrate by vacuum evaporation and the surface was annealed by hydrogen flame to generate the flat polycrystalline Au surface. This procedure is necessary for the effective excitation of SP as well as for the effective immobilization of antibodies on the surface. This Au film on the glass substrate was then attached on a glass prism with an index-matching oil, illuminated by visible or near-IR light, and the reflectivity is recorded upon changing the incident angle. The resulting plot of the reflectivity against the incident angle, ‘SPR curve’, has a sharp and deep valley at the specific angle defined as the ‘SPR angle’. Since the SPR angle sensitively increases with an increase in the dielectric constant of the molecules on the surface, which is consistent with the amount of bound analyte to the ligand immobilized on the surface, this SPR phenomenon can be employed as an effective method of LBA.
Based on this principle of an SPR biosensor, we have developed the selective heating of a single antibody capturing the target pathogenic particle, as summarized in the Scheme. At the SPR angle, the almost incident light energy is absorbed by the gold film, transferred to the SPs, and finally converted to heat. Practical SPR biosensor systems employ a dim excitation light because the resulting heat disturbs the detection of ligand-analyte binding. In contrast, we intentionally employ a high-power near-IR laser as a light source to generate enough heat to drive the DNA polymerase upon the enhancement process. The pathogen-specific antibody (‘test’) and the antibody lacking the binding ability (‘control’) are linked with the primer sequences for the RCA with the linker molecules and then immobilized on the Au film as 1 mm ‘test’ and ‘control’ spots, respectively.
The SPR angle increases with increasing amount of the bound analyte as described above. Accordingly, when we illuminate the high-power near-IR laser at an incident angle (q), greater than the SPR angle for the antibodies themselves (q), the SP fields should be mainly generated at the pathogen-bound antibodies, and the resulting heat can induce DNA elongation from the primers immobilized on the antibodies binding the pathogens. The amplified DNA (amplicons) elongating around the pathogen-bound antibodies contributes more effectively to the increase in the SPR angle than the initial binding of pathogens to the antibodies. Here, RCA not only elongates amplicons linearly, but also introduces many branches on them, using another primer sequence on the amplicons. As a result, accelerating heat generation leading to further acceleration of DNA elongation around the pathogen-bound antibody can be expected. The resulting pathogen wrapped by the amplicon can be easily detected and counted after fluorescence staining. The extreme difference in the efficiency of the DNA elongation between the ligand and control spots should be particularly effective for the digital detection of pathogens suppressing the influences of weak background interactions (Scheme 2).
To confirm the validity of this assay, we carried out an experiment using 3-μm IgG-conjugated latex beads as a model of the pathogen. Anti-IgG and anti-BSA (control) antibodies were modified with 5’-NH2-M13M4-primers. A 6-mm silicon rubber through-hole well was then attached to a 50-nm gold film on a coverslip. Both the antibody conjugates were immobilized on the gold surface to form 1-mm ‘test’ and ‘control’ spots, respectively. Following immuno-reaction with the pathogen models, the surface was immersed in an RCA solution (comprising M13mp18, Bst polymerase, and dNTPs), and illuminated by a p-polarized 808-nm 600-mW laser light for 30-60 min through a glass prism at the incident angle (q’’). This offset incident angle (dqoffset=q’’-q) is the expected SPR angle for the antigen-bound antibodies covered with a sufficient amount of the amplicons. The resulting amplicons were stained with SYBR Green I and observed via confocal microscopy (~40x magnification). The bright fluorescence spots were observed specifically at the test spot. The locations of the fluorescence spots were consistent with those of the particles in the transmission image at higher magnification. This result indicates that site-selective SP heating successfully induced RCA at the binding site of the 3-μm latex beads. Although clear imaging and counting of the 3-μm model pathogens can be demonstrated, the size is consistent with the assay of the pathogenic bacteria.
The most significant and urgent target pathogens are, in fact, the virus having much smaller sizes (approximately 100 nm) than that of the model pathogens tested above.
When our method is applied to the virus assay, the main concern is the decrease in the contrast between the fluorescence intensities of the virus-bound and the free antibodies. The change in the SPR angle upon binding the virus by the antibody (dq=q’-q) should be smaller than that for the bacteria. Accordingly, the difference between the local reflectances at the immobilization spot of the virus-bound antibodies and that of the free antibodies at the SPR angle for the virus-bound antibodies covered with the amplicons (q’’) may decrease, leading to a small difference in the amplicon yields at each site corresponding to the heat generation efficiencies. Thus, setting the offset angle (θoffset=q’’-q) will likely be crucial during the practical application of this method.
Since our concept depends on site-selective heating at the single-molecule scale, the optimization of the illumination angle should be based not on the SPR measurements of the bulk Au surfaces but on the estimation of the SP excitation efficiency at the single-molecule scale on the surface. We then performed the estimation by employing a 100 nm latex particle as the model of the virus and monitoring the intensities of the scattering light from the model virus by surface imaging as the parameter of the SP field intensity around the antibody-bound virus. Although the bulk SPR curve did not show an obvious change after the binding of the model virus, the bright scattering spots corresponding to the model virus were observed at a specific angle, indicating the SPR angle at the virus-bound antibody. Using this optimization method of the illumination angle, we may prospect the application of the present method to virus assays (Figure 11).
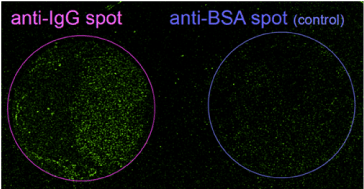
Figure 11. Multispot fluorescence assay based on SP-heating enhanced RCA on gold surface; a) anti-IgG immobilized test spot, b) anti-BSA immobilized control spot; both 1mm spots were immersed in the solution of primer-bound IgG for 30 min and SP heating was performed in RCA solution; after fluorescence staining by SYBR-Green I confocal imaging was performed by 488 nm excitation using a x40 objective lens.
We have described the recent progress of the methods for pathogen assays and their applications to practical diagnostic devices. Both the methods, the genome base and the antibody base, have remarkably progressed by the incorporation of novel concepts such as digital detection and the application of SP as a nano-scaled heating method, and new models of diagnostic devices with specific features have been launched. However, each method does not seem to achieve a satisfactory outcome as stand-alone at present as some of the properties originating from the assay concept are in tradeoff relation. Therefore, the combined use of both the methods to complement the benefits should be effective and necessary for the infection control of COVID-19 and the new EIDs.
We hope that this review will be useful not only for healthcare professionals to improve the accuracy and throughput of the routine assays, but also for the representatives of diagnostic equipment at the institutions, considering the purchase of next-generation assay devices by understanding their principles and features.
View Supplementary Data
- Alahi EE, Mukhopadhyay SC (2017) Detection methodologies for pathogen and toxins: A review. Sensors (Switzerland) 17: 1-20. [Crossref]
- Carter LJ (2020) Assay techniques and test development for COVID-19 diagnosis. ACS Cent Sci 6: 591-605. [Crossref]
- Watson J, Whiting PE, Brush JE (2020) Interpreting a covid-19 test result. BMJ 369: 1-7. [Crossref]
- Black JRM, Bailey C, Przewrocka J, Dijkstra KK, Swanton C (2020) COVID-19: the case for health-care worker screening to prevent hospital transmission. Lancet 395: 1418-1420. [Crossref]
- Lescure FX (2020) Clinical and virological data of the first cases of COVID-19 in Europe: a case series. Lancet Infect Dis 20: 697-706.
- Han MS, Byun JH, Cho Y, Rim JH (2020) RT-PCR for SARS-CoV-2: quantitative versus qualitative. Lancet Infect Dis 3099: 30424.
- Böhmer MM (2020) Investigation of a COVID-19 outbreak in Germany resulting from a single travel-associated primary case: a case series. Lancet Infect Dis 20: 920-928.
- Kucirka LM, Lauer SA, Laeyendecker O, Boon B, Lessler J (2020) Variation in False-Negative Rate of Reverse Transcriptase Polymerase Chain Reaction–Based SARS-CoV-2 Tests by Time Since Exposure. Ann Intern Med 173: 262-267. [Crossref]
- Wikramaratna P, Paton RS, Ghafari M, Kingdom U (2020) Estimating false-negative detection rate of SARS-CoV-2 by RT-PCR 413: 0-13.
- FAO/WHO (2012) Proposed draft guidelines on the application of general principles of food hygiene to the control of viruses in food. codex alimentarius commission: Report of the forty-third session of the codex committee on food hygiene 12: 46-63.
- Sumio Shinoda SM, Tamaki Mizuno (2019) General review on hog cholera (classical swine fever), african swine fever, and salmonella enterica serovar choleraesuis infection. J Disaster Res 14: 1105-1114.
- Blome S, Staubach C, Henke J, Carlson J, Beer M (2017) Classical swine fever-an updated review. Viruses 9: 1-24. [Crossref]
- Ribbens S, Dewulf J, Koenen F, Laevens H, De Kruif A (2004) Transmission of classical swine fever. A review. Vet Q 26: 146-155. [Crossref]
- Sellwood C, Asgari-Jirhandeh N, Salimee S (2007) Bird flu: If or when? Planning for the next pandemic. Postgrad Med J 83: 445-450.
- Thomas JK, Noppenberger J (2007) Avian influenza: A review. Am J Heal Pharm 64: 149-165.
- Myers KP, Olsen CW, Gray GC (2007) Cases of swine influenza in humans: A review of the literature. Clin Infect Dis 44: 1084-1088. [Crossref]
- Ligon BL (2005) Avian influenza virus h5n1: A review of its history and information regarding its potential to cause the next pandemic. Semin Pediatr Infect Dis 6: 326-335.
- Shirato K (2020) Development of genetic diagnostic methods for detection for novel coronavirus 2019(nCoV-2019) in Japan. Jpn J Infect Dis 73: 304-307. [Crossref]
- Shirato K, Nao N, Matsuyama S, Kageyama T (2020) Ultra-rapid real-time RT-PCR method for detecting middle east respiratory syndrome coronavirus using a mobile PCR device, PCR1100. Jpn J Infect Dis 73: 181-186.
- Okamaoto K (2020) An assessment of real-time RT-PCR kits for SARS-CoV-2 detection. Jpn J Infect Dis. [Crossref]
- Kontou PI, Braliou GG, Dimou NL, Nikolopoulos G, Bagos PG (2020) Antibody tests in detecting SARS-CoV-2 infection: A meta-analysis. Diagnostics 10: 1-5.
- Waghmare A (2020) Reliability of self-sampling for accurate assessment of respiratory virus viral and immunologic kinetics. medRexiv 1: 1-10.
- Champlot S, Berthelot C, Pruvost M, Andrew Bennett E, Grange T, et al. (2010) An efficient multistrategy DNA decontamination procedure of PCR reagents for hypersensitive PCR applications. PLoS One 5: 1-9.
- Miller JJ, Levinson SS (1996) Interferences in immunoassays, in Immunoassay. Diamandis EP, Christopoulos TK, (Eds). London: Academic press pp.165-190.
- Butler JE (1996) Solid phases in immunoassay in Immunoassay. Diamandis EP, Christopoulos TK (Eds). London: Academic press, Inc pp.205-226.
- Butler JE (2004) Solid supports in ELISA and other assays in Molecular Diagnosis of Infectious Diseases, 2nd edition, Decker J, Reischl U (Eds). Totowa: Humana Press, pp.333-372.
- Chang T, Wu J, Chang J (2020) Fighting COVID-19: Integrated micro- and nanosystems for viral infection diagnostics. Matter 3: 1-24.
- Quan PL, Sauzade M, Brouzes E (2018) DPCR: A technology review. Sensors (Switzerland)18: 1271. [Crossref]
- Pecoraro S (2019) Overview and recommendations for the application of digital PCR.
- Cao Y, Yu M, Dong G, Chen B, Zhang B (2020) Digital PCR as an emerging tool for monitoring of microbial biodegradation. Molecules 25: 1-10.
- Biorad (2018) Droplet digital TM PCR droplet digital TM PCR applications guide. Biorad p. 145.
- Ullerich L, Campbell S, Krieg-Schneider F, Bürsgens F, Stehr J (2017) Ultra-fast PCR technologies for point-of-care testing. Laboratoriums Medizin 41: 239-244.
- Fischer SK (2015) Emerging technologies to increase ligand binding assay sensitivity. AAPS J 17: 93-101. [Crossref]
- Wilson DH (2016) The Simoa HD-1 analyzer: A novel fully automated digital immunoassay analyzer with single-molecule sensitivity and multiplexing. J Lab Autom 21: 533-547. [Crossref]
- Cohen L, Walt DR (2017) Single-molecule arrays for protein and nucleic acid analysis. Annu Rev Anal Chem 10: 345-363.
- Bruschini C, Homulle H, Antolovic IM, Burri S, Charbon E (2019) Single-photon avalanche diode imagers in biophotonics: review and outlook. Light Sci Appl 8: 1-10.
- Ju H, Lai G, Yan F (2017) Electrochemiluminescent immunosensing,” in Immunosensing for Detection of Protein Biomarkers. (Eds) Amsterdam: Elsevier pp,171-206.
- Roche (2020) Elecsys Anti-SARS-CoV-2 Elecsys Anti-SARS-CoV-2. pp. 1-7.
- Innis MA, Gelfand DH (1990) Optimization of PCRS in PCR Protocols A Guide to Methods and Applications. Innis MA, Gelfand DH, Sninsky JJ, White TJ (Eds) San Diego: Academic press Inc.
- Basha IHK, ETW Ho, CM Yousuff, NH Bin Hamid (2017) Towards multiplex molecular diagnosis-A review of microfluidic genomics technologies. Micromachines 8: 266. [Crossref]
- Oda RP (1998) Infrared-Mediated thermocycling for ultrafast polymerase chain reaction amplification of DNA. Anal Chem 70: 4361-4368. [Crossref]
- Giordano BC, Ferrance J, Swedberg S, Hühmer ARF, Landers JP (2001) Polymerase chain reaction in polymeric microchips: DNA amplification in less than 240 seconds Anal Biochem 291: 124-132. [Crossref]
- Kristin A Hagan, Reedy CR, Uchimoto ML, Basu D, Engel DA, et al. (2011) An integrated, valveless system for microfluidic purification and reverse transcription-PCR amplification of RNA for detection of Infectious Agents. Lab Chip 11: 957-961.
- Phaneuf CR (2015) Thermally multiplexed polymerase chain reaction. Biomicrofluidics 9: 1-13. [Crossref]
- Liu W, Warden A, Sun J, Shen G, Ding X (2018) Simultaneous detection of multiple HPV DNA via bottom-well microfluidic chip within an infra-red PCR platform. Biomicrofluidics 12: 1-10.
- Liu W, Zhang M, Liu X, Sharma A, Ding X (2020) A Point-of-Need infrared mediated PCR platform with compatible lateral flow strip for HPV detection. Biosens Bioelectron 96: 213-219. [Crossref]
- Yu Y, Li B, Baker CA, Zhang X, Roper MG (2012) Quantitative polymerase chain reaction using infrared heating on a microfluidic chip. Anal Chem 84: 2825-2829.
- Jenny A (2020) From sample to PCR product in under 45 minutes: a polymeric integrated microdevice for clinical and forensic DNA analysis. Lab Chip 13: 1384-1393. [Crossref]
- Kim H, Vishniako S, Faris GW (2009) Petri dish PCR: laser-heated reactions in nanoliter droplet arrays. Lab Chip 9: 1230-1235.
- Thermo Fisher Scientific (2020) Realtime PCR handbook.
- Keer JT (2008) Quantitative Real-time PCR Analysis,” in Essentials of Nucleic Acid Analysis: A Robust Approach, J. T. Keer and L. Birch, Eds. Cambridge: The Royal Society of Chemistry pp; 132-166.
- Hindson CM (2013) Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods 10: 1003-1005. [Crossref]
- Maheshwari Y, Selvaraj V, Hajeri S, Yokomi R (2017) Application of droplet digital PCR for quantitative detection of Spiroplasma citri in comparison with real time PCR. PLoS One 12: 1-16.
- Basu AS (2017) Digital Assays Part I: Partitioning Statistics and Digital PCR. SLAS Technol 22: 369-386.
- Majumdar N, Banerjee S, Pallas M, Wessel T, Hegerich P (2017) Poisson Plus Quantification for Digital PCR Systems. Sci Rep 7: 1-10
- Raether H (1998) Surface Plasmons on Smooth and Rough Surfaces and on Gratings. Berlin: Springer-Verlag.
- Chatterjee DK, Diagaradjane P, Krishnan S (2011) Nanoparticle-mediated hyperthermia in cancer therapy. Ther Deliv 2: 1001-1014.
- Jain PK, Huang X, El-Sayed IH, El-Sayed M (2008) Noble metals on the nanoscale: optical and photothermal properties and some applications in imaging, sensing, biology, and medicine. Acc Chem Res 41: 1578-86
- Son JH (2015) Ultrafast photonic PCR Light Sci Appl 4: 7
- Ishida A, Majima T (1999) Surface plasmon enhanced fluorescence spectroscopy towards observation of molecular exchange in a self-assembly monolayer. Chem Comm 1299-1300.
- Ishida A, Majima T (2000) Surface plasmon enhanced fluorescence measurement on flat and constructed gold surfaces. The Analyst 125: 535-540
- Geddes CD (2010) Metal-Enhanced Fluorescence. Hoboken: John Wiley & Sons.
- Matherly D (2013) Trick of the light, Nature 495: 58-59.
- Willets KA, Van Duyne RP (2007) Localized surface plasmon resonance spectroscopy and sensing. Annu Rev Phys Chem 8: 267-297.
- Rahman M (2014) Nanoplasmonics and its applied devices Jnsm 1: 1-15.
- Mohamed MB, Volkov V, Link S, El-sayed MA (2000) The lightning gold nanorods: fluorescence enhancement of over a million compared to the gold metal. pp.17-523.
- He J (2013) Practical Guide to elisa development in the immunoassay handbook: theory and applications of ligand binding, elisa and related techniques, 4th ed., D. Wild, R. John, C. Sheehan, S. Binder, and J. He, Eds. Oxford: Elsevier. pp. 381-394.
- Wei X (2006) A specific picomolar hybridization-based ELISA assay for the determination of phosphorothioate oligonucleotides in plasma and cellular matrices. Pharm Res 23: 1251-1264. [Crossref]
- Fu Q, Zhu J, Van Eyk JE (2010) Comparison of multiplex immunoassay platforms. Clin Chem 56: 314-318.
- Matsukuma E (2006) Development of fluorescence-linked immunosorbent assay for high throughput screening of interferon-γ. Allergol Int 55: 49-54.
- Ahmed S (2020) Current advances in immunoassays for the detection of antibiotics residues: a review. Food Agric Immunol 31: 268-290.
- Sigma Millipore (2017) SMCxPRO immunoassay system sensitivity you can count on.
- Comley J (2012) Elisa assays recent innovations take analyte.
- Gibbs J, Vessels M, Rothenberg M (2017) Selecting the Detection System - Colorimetric, Fluorescent, Luminescent Methods for ELISA Assays, ELISA Tech. Bull. Corning Life Sci 5: 1-14.
- Chen D (2018) Comparison of chemiluminescence immunoassay, enzyme-linked immunosorbent assay and passive agglutination for diagnosis of Mycoplasma pneumoniae infection. Ther Clin Risk Manag 14: 1091-1097. [Crossref]
- Xu L, Burkin M, Eremin S, Dias ACP, Zhang X (2019) Development of competitive ELISA and CLEIA for quantitative analysis of polymyxin B. Food Anal Methods 12: 1412-1419.
- Seto Y, Iba T, Abe K (2001) Development of ultra‐high sensitivity bioluminescent enzyme immunoassay for prostate‐specific antigen (PSA) using firefly luciferase. J Biol Chem Lumin 16: 285-290. [Crossref]
- Sakamaki N (2012) Bioluminescent enzyme immunoassay for the detection of norovirus capsid antigen. Clin Vaccine Immunol.19: 1949-1954. [Crossref]
- Ren W (2019) One-Step ultrasensitive bioluminescent enzyme immunoassay based on nanobody/nanoluciferase fusion for detection of aflatoxin B 1 in Cereal. J Agric Food Chem 67: 5221-5229 [Crossref]
- Liang M (2007) Detection of high- and low-affinity antibodies against a human monoclonal antibody using various technology platforms. ASSAY Drug Dev Technol 5: 655-662. [Crossref]
- Prieto B, Miguel D, Costa M, Coto D, Álvarez FV (2010) New quantitative electrochemiluminescence method (ECLIA) for interleukin-6 (IL-6) measurement. Clin Chem Lab Med 48: 835-838.
- Zhang N, Zhang R, Yang X, Qi H, Zhang C (2019) Recent advances in electrogenerated chemiluminescence biosensing methods for pharmaceuticals. J Pharm Anal 9: 9-19.
- Qi H, Zhang C (2020) Electrogenerated chemiluminescence biosensing. Anal Chem 92: 524-534.
- Woodbury N, Bald E, Geist B, Yang TY (2019) Application of multiplexed pharmacokinetic immunoassay to quantify in vivo drug forms and coadministered biologics. Bioanalysis 11: 2251-2268.
- Cohen L (2020) Single Molecule Protein Detection with Attomolar Sensitivity Using Droplet Digital Enzyme-Linked Immunosorbent Assay. ACS Nano.
- Kupcova SH, Cizkova J, Cervenka J, Vodicka P (2017) Advances in proteomic techniques for cytokine analysis: focus on melanoma research. Int J Mol Sci 18: 1-10. [Crossref]
- Kawahara Y, Ishida A (2016) Surface plasmon resonance (SPR) sensing based on DNA elongation by site-selective surface plasmon (SP) field heating toward ultra-high sensitive detection of single pathogenic particles. in ECS Transactions 75: 1-10.