An organometallic arene ruthenium (II) compound comprising a 1,1-bis(diphenylphosphino)methane (dppm) ligand, [Ru(p-cymene) (dppm)Cl2] has been reported to exhibit interesting anticancer activity in breast cancer cells. However, its underlying molecular mechanism of action remains largely unexplored. We investigated the anticancer activity and BRCA1 inhibition of the ruthenium compound in BRCA1-associated breast cancer cells and a high degree of cytotoxicity was displayed against all selected BRCA1-associated breast cancer cells. Triple-negative BRCA1-deficient HCC1937 cells were significantly more sensitive toward the ruthenium compound than triple-negative BRCA1 wild-type MDA-MB-231 and BRCA1 wild-type MCF-7 cells. The anticancer activity of the ruthenium compound was more potent than the activity of the dppm ligand alone and cisplatin against all three cell lines. The difference in cytotoxicity was associated with the uptake and accumulation of ruthenium atoms, cell cycle progression, and the generation of apoptotic cells and the resulting increase in the expression of p53 and p21 mRNAs along with their respective proteins. In a PCR-based assay, the ruthenium compound inhibited amplification of the BRCA1 gene in all three cell lines, especially in HCC1937 cells. However, the expression of BRCA1 mRNA in the cell lines was regulated differently. Expression of the BRCA1 protein in the three cell lines was reduced in HCC1937 > MDA-MB-231 > MCF-7. The findings of this work provide insights into the molecular mechanism of action of [Ru(p-cymene) (dppm)Cl2] in BRCA1-associated breast cancer cells.
anticancer activity, apoptosis, cell cycle, Ru-dppm compound, triple negative breast cancer
Globally, breast cancer is the second leading cause of cancer-related deaths in women [1]. Breast cancer is a heterogeneous disease that is classified into five main intrinsic molecular subtypes based on differences in gene expression profiling. These subtypes are known as luminal A, luminal B, human epidermal growth factor receptor 2 (HER2) overexpression, basal-like, and normal-like [2,3]. Triple-negative breast cancer (TNBC), which accounts for approximately 15% of breast cancers, is characterized by a lack of expression of the estrogen receptor (ER), the progesterone receptor (PR), and HER2 [4]. TNBC has recently gained clinical importance and become the subject of intense investigation. Its aggressive phenotype, high metastatic progression, poor prognosis, earlier recurrence and shorter survival distinguish it from other breast cancer subtypes [5]. TNBC is overlapped up to 70% by the basal-like subgroup [2,6]. Currently, there is no standard cancer drug treatment regimen nor specific targeted therapy for TNBC patients that is effective. However, the recently acquired better understanding of the molecular basis of different breast cancer subtypes and mutations within TNBC has allowed the exploration of some innovative targeted therapies, bringing new hope for TNBC patients. Some promising targeted therapies such as poly (ADP-ribose) polymerase (PARP) inhibitors and platinum-based drugs are currently being assessed in clinical trials [7,8].
Emerging data have revealed that approximately 20-30% of TNBC patients harbor germline mutations in the BRCA1 gene [9,10]. The BRCA1 protein has multiple functions that help maintain genomic stability in cells. It is involved in DNA damage repair, cell cycle check-point control, regulation of transcriptional activities, and ubiquitination. However, defects in the BRCA1 protein led to a failure of its functions [7]. It has been reported that some TNBC patients with germline BRCA1 mutations achieved pathological complete responses (pCRs) after treatment with platinum-based neoadjuvant chemotherapy [11-14]. In addition, treatment with a combination of carboplatin and anthracycline/taxane significantly improved the rate of pCR among TNBC patients [15]. Consequently, several studies focused on modifying standard chemotherapeutic regimens using new chemotherapeutic agents with significantly pCR rates [11]. Following clinical trials, a new paradigm was proposed for the treatment of TNBC patients that made use of dysfunctional BRCA1 proteins or a specific BRCA1-mediated homologous recombination (HR) repair defect [7,16]. A report was published that breast cancer cells with dysfunctional BRCA1 were sensitive to chemotherapeutic agents [17]. BRCA1-deficient HCC1937 cells were more sensitive than BRCA1-wild type MCF-7 cells to the platinum drug cisplatin, but HCC1937 cells in the presence of exogenous BRCA1 exhibited resistance to cisplatin [18]. However, the antisense inhibition of BRCA1 in cisplatin resistant breast cancer cells resulted in increased sensitivity to cisplatin, a reduced efficacy of DNA repair, and an increased rate of apoptosis [19,20]. Furthermore, the inhibition of endogenous BRCA1 expression in ovarian cancer cells resulted in an increased sensitivity to platinum chemotherapy [21]. Various chemotherapeutic regimens have been used to improve the treatment of each breast cancer subtype [22,23] but chemotherapeutic treatment can be more complicated when breast cancer patients have functional BRCA1 protein. As a result, the profiling of BRCA1 protein expression has been referred as a novel biomarker to predict the outcome and efficacy of treatment among breast cancer patients [24,25].
The use of platinum drugs, whether alone or combined, has increasingly encountered clinical limitations in the treatment of TNBC. It has been reported that many breast cancer patients who initially responded well to anticancer platinum drugs later became drug-resistant [26-29]. The primary limitation of platinum drugs has been their adverse side effects, and more lately, the induction of secondary mutations in the BRCA1 gene leading to acquired cellular resistance [30-33]. Over decades, a series of potent and efficient metal-based compounds have been developed to combat the problems associated with the use of platinum-based drugs and ruthenium(II) compounds are the metal-based compounds of choice for this purpose [34]. A rational drug design aimed at increasing the potency of anticancer ruthenium compounds should be based on its chemical structure and activity, ligand exchanges, oxidation states and iron mimic activity [35]. The transition metal ruthenium plays a vital role in metal transport by means of passive diffusion and a transferrin-dependent mechanism [36]. Numerous organometallic ruthenium-based compounds have been synthesized and developed as anticancer agents to overcome the clinical limitations of cisplatin [35,37]. A promising series of ruthenium compounds presented a half-sandwich with piano stool geometry in the following structure [(η6)-arene Ru(X)(Y)(Z)] [38-40].
In organometallic ruthenium (II)-arene complexes, the hydrophilicity of the metal center, coordinated to a hydrophobic p-cymene ligand, contributed to the excellent cytotoxicity [41-43]. A distorted tetrahedral piano-stool geometry coordinated to the ruthenium (II) center with p-cymene, a chloro ligand, and a ligand with phosphorus donors proved to be an interesting complex [44,45]. In addition, ruthenium compounds have been reported to regulate the
biomarker genes that were involved in the apoptotic pathway (p53), cell cycle progression (p21) and the homologous recombination repair gene BRCA1 [8]. The RAPTA-EA1 compound reduced BRCA1 replication significantly more in BRCA1-deficient HCC1937 cells than in BRCA1-proficient MCF-7 cells [8,46], and the expression of BRCA1 mRNA was upregulated in HCC1937 cells in the presence of the ruthenium compound but downregulated in the MCF-7 cells. However, the production of the BRCA1 protein was suppressed in the tested cells. In this study, BRCA1 mRNA expression in HCC1937 cells was not proportionally correlated to its respective protein expression as it was in previous studies. This inverse correlation between BRCA1 mRNA and its protein level could result from molecular distinctions between subtypes of breast cancers [47].
Recently, ruthenium(II)-arene compounds bearing a chelating ligand of 1,1-bis(diphenylphosphino)methane (dppm), termed [Ru(p-cymene)(dppm)Cl2], have been evaluated as anticancer agents. In a study that compared the anticancer activities of a ruthenium(II) complex and free ligands, the complex performed better against cell proliferation on A2780 and HT-29 cell lines, giving IC50 values of 0.7 and 0.6 µM , respectively [48]. These results suggest that the ruthenium metal prevented the ligands being oxidized before they could interact with the biological target [49]. The complex also inhibited cell growth of several human cancers through multi-mechanisms, such as interference with DNA replication, induction of cell cycle arrest, downregulation of anti-apoptotic genes and upregulation of pro-apoptotic genes that ultimately trigger the programmed cell death pathway [50]. Our preliminary study of the proposed ruthenium (II)-arene compound containing a dppm ligand (Figure 1) revealed a significant anti-proliferative activity against some selected breast cancer cell lines. It was found to be highly active against breast cancer cell lines with cytotoxicity properties comparable to cisplatin [51]. So far, the underlying molecular mechanism of action of [Ru(p-cymene)(dppm)Cl2] on breast cancer cells remains largely unexplored. In this study, therefore, we investigated the induction by [Ru(p-cymene)(dppm)Cl2] of anticancer activity and BRCA1 inhibition in triple-negative BRCA1-deficient HCC1937, triple-negative BRCA1 wild-type MDA-MB-231, and sporadic BRCA1 wild-type MCF-7 breast cancer cells.
Figure 1. Chemical structure of [Ru(p-cymene)(dppm)Cl2]
Compound: The η6-arene-based ruthenium compound comprised a 1,1-bis(diphenylphosphino)methane (dppm) ligand, giving the compound [Ru(p-cymene)(dppm)] (Figure 1). The dppm ligand was kindly provided for this study by Asst. Prof. Dr. Nararak Leesakul, Division of Physical Science and Center of Excellence for Innovation in Chemistry, Faculty of Science, Prince of Songkla University. The preparation of the [Ru(p-cymene)(dppm)Cl2] complex and its dppm ligand was described previously [51].
Human breast cancer cell lines and culture conditions: The human breast cancer cell lines MCF-7 (BRCA1-competent), triple-negative MDA-MB-231 (BRCA1 wild-type), and triple-negative HCC1937 (BRCA1-deficient, 5382insC), were obtained commercially from American Type Culture Collections (ATCC, Rockville, MD). All cell lines were cultured in the cell growth medium as a monolayer. MCF-7 and MDA-MB-231 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) and HCC1937 cells were maintained in Roswell Park Memorial Institute 1640 medium (RPMI 1640). Supplements of 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin were both added to each cell culture medium. All cells were maintained in a humidified incubator at 37 °C in 5% carbon dioxide (CO2).
MTT proliferation assay: MTT reduction by metabolically viable cell activity was used to initially investigate the cytotoxic effects of [Ru(p-cymene) (dppm)Cl2] and the dppm ligand alone on the cell viability of the selected breast cancer cell lines. A logarithmically monolayer cells were grown in cell culture flasks. Cell pellets were collected by trypsinization and centrifugation at 500 × g for 5 min at 25 °C and flat-bottomed 96-well microplates were seeded with 100 μL per well of a single cell suspension of 104 cells in a complete medium. To allow cell attachment, the plates were incubated overnight in a humidified incubator at 37 °C in 5% CO2. When cell growth reached approximately 80% confluence, the medium was removed and fresh medium added containing 200 mL of either [Ru(p-cymene) (dppm)Cl2] or the dppm ligand, at final concentrations of 0, 0.01, 0.1, 1, 5, 10, 25, 50, and 100 µM. The medium was removed after 48 h of incubation and cells were then washed twice with 100 mL of phosphate bufferred saline (PBS). Subsequently, 100 μL of 0.5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) solution was added to each well and the plates were incubated for a further 4 h at 37 °C in 5% CO2. After incubation, the solution was aspirated and 200 mL of dimethylsulfoxide solution (DMSO) was added to dissolve the formazan crystals formed. The solution was then spectrophotometrically measured at 570 nm. To compare the sensitivity of the studied cells toward the growth inhibitory potential of the dppm ligand and the [Ru(p-cymene) (dppm)Cl2] complex, the absorbance data were totaled, and cell viability was calculated from the following equation (1):
Cell viability (%) = ((absorbance of the treated wells)/(absorbance of the vehicle-treated control wells)) x 100 (1)
Percentage cell viability was plotted against various concentrations of the ruthenium compound to determine the 50% inhibiting concentration (IC50), defined as the concentration of the ruthenium compound or dppm ligand which inhibited cell viability by at least 50% compared to the control condition.
Subcellular ruthenium contents distribution: The subcellular distribution of ruthenium atoms in the breast cancer cell lines was investigated using ICP-MS. Cancer cells at a density of 5×105 cells/mL were placed in a 75 cm3 cell culture flask and incubated for 48 h at 37 °C in 5% CO2 with [Ru(p-cymene) (dppm)Cl2] at the IC50 and 0.1% DMSO control. After 48 h, cells were rinsed twice with PBS, 0.25% trypsin/EDTA was added and the cells were centrifuged at 1,000 × g for 10 min at 4 °C. Cellular fractions were produced as described previously [52,53]. Briefly, cell pellets were lysed with a sucrose-mannitol medium and the cell homogenates were separated into subcellular components by centrifugation at 1,000 × g for 10 min at 4 °C. Crude nuclear sediment and crude cytoplasmic solution were collected separately. The crude nuclear sediment was resuspended in a Tris-Mg-NaCl buffer made up of 250 mM sucrose in a Tris-Mg-NaCl buffer and 2 volumes of 2.3 M sucrose and centrifuged at 40,000 × g for 30 min. The nuclear sediment was then resuspended in a Tris-Mg-NaCl buffer containing 250 mM sucrose, 0.1% Triton X-100 (w/w) and centrifuged at 1,000 × g for 10 min. After centrifugation, the sediment and the supernatant were separated. The collected sediment was resuspended in a 1% sodium lauryl sulfate (SDS) solution (150 mM NaCl, 10 mM Tris-Cl (pH 8.0), 1 mM EDTA) and kept at room temperature overnight. The collected supernatant was centrifuged at 17,000 × g for 30 min and the sediment produced was dissolved in double-distilled water to obtain the nuclear fraction. The supernatant was then transferred and centrifuged at 12,000 × g for 10 min to yield the solution containing the cytoplasmic fraction and the mitochondrial pellet. The cytoplasmic fraction was collected. The mitochondrial pellet was resuspended in a homogenization medium of sucrose-mannitol and the suspension was centrifuged at 1,000 × g for 5 min. The formed precipitate was discarded. The supernatant was collected and resedimented by centrifugation at 12,000 × g for 10 min to produce a mitochondrial pellet. The mitochondrial pellet was dissolved in PBS buffer and stored at -20 °C. The three isolated subcellular fractions (cytoplasmic, mitochondrial, and nuclear fractions) were later analyzed to determine ruthenium contents using an inductively coupled-plasma mass spectrometer (ICP-MS). Each experiment was conducted in duplicate.
Cell cycle distribution analysis: To assess cell cycle distribution, cellular DNA contents were determined by flow cytometry. Cells were seeded into 6-well culture plates at 106 cells/mL and grown at 37 °C in 5% CO2. At approximately 80% monolayer cell growth confluence, the medium was removed and treated for 48 h with the IC50 of the ruthenium compound. Cells were then trypsinized, rinsed twice with 0.5 mL of cold PBS, and centrifuged at 300 × g for 5 min. The cells were then fixed with cold 70% ethanol and stored at -20 °C overnight. The fixed cells were washed twice with cold PBS. Afterward, 1 mL staining solution of PBS containing 100 µg/mL of RNase A, 50 µg/mL of propidium iodide (PI), and 0.1% of Triton X-100 was added to the cell pellets, which were then resuspended and incubated at 37 °C for 30 min in darkness. Finally, the fluorescence of 20,000 cells was detected using a FACSCanto flow cytometer (BD Biosciences) and the percentage cell population was analyzed with MultiCycle software. DNA content histograms were produced based on the percentage of cells in the G0/G1, S, and G2/M phases.
Annexin V/PI double staining apoptotic detection by flow cytometer: The double staining Annexin V-FITC and PI staining detection kits (Invitrogen) were used to detect apoptotic and necrotic cells using flow cytometry following the manufacturer's protocol. Briefly, following incubation with the IC50 of the ruthenium compound, the untreated and treated cells were trypsinized, rinsed twice with 0.5 mL of cold PBS, and centrifuged at 500 × g for 5 min. The supernatant was discarded and the cell pellets were resuspended in 100 µL of 1xAnnexin-binding buffer. Then, 5 μL of Alexa Fluor 488, and 1 μL of 100 μg/mL PI solution were gently mixed into each cell suspension. The suspensions were then incubated at room temperature for 15 min in darkness. After incubation, 400 μL of 1xAnnexin-binding buffer were added and mixed gently. The Annexin V binding staining of 20,000 cells was immediately analyzed using FACS (fluorescence activated cell sorting) flow cytometry (FACSCanto, BD Biosciences), measuring emissions at 530 and 575 nm from an excitation wavelength of 488 nm.
Determination of cellular DNA damage by semi-quantitative polymerase chain reaction (QPCR): Cells were exposed for 48 h to various doses of [Ru(p-cymene)(dppm)Cl2] at 37 °C in 5% CO2. The cell pellets were collected by trypsinization and centrifuged at 500 × g for 5 min. The cellular genomic DNA of the cells was isolated using a procedure from a previous study [53]. The BRCA1 exon 11 fragment (3426-bp) was the cellular DNA target that identified the effect of the ruthenium compound on DNA amplification by PCR [53]. Briefly, the PCR reaction mixture consisted of Ru-treated genomic DNA template (400 ng), Phusion Hot Start DNA polymerase (2 units), MgCl2 (1.5 mM), 1xPhusion™ GC Buffer, dNTPs (300 μM of each), forward primer (5/-GCCAGTTGGTTGATTTCCACC-3/), reverse primer (5/-GTAAAATGTGCTCCCCAAAAGC-3/) (0.5 μM of each primer), and double distilled water to a final volume of 50 μL. PCR was carried out in triplicate under the following cycling condition: 1 cycle of an initial step at 94 °C for 3 min, followed by 30 cycles of 94 °C for 30 s, 60 °C for 45 s, 72 °C for 2 min, and a final extension at 72 °C for 7 min. The targeted PCR products were then electrophoresed on a 1% agarose gel at 100 V for 60 min. The agarose gel was stained with ethidium bromide and illuminated under UV light. The inhibition of DNA polymerization by ruthenation was assessed using semi-quantitative polymerase chain reaction (QPCR). The band intensity of DNA amplification products was measured using a Bio-Rad Molecular Imager with the Molecular Dynamics program (version 1.0.2, Bio-Rad, Hercules, CA, USA, 1994). The percentage of DNA amplification was plotted against various concentrations of the ruthenium compound. The lesion frequency per strand of the 3,426 bp of the BRCA1 exon 11 fragment was determined by the Poisson equation (2) [54],
S = -ln Ad/A (2)
where S is the lesion frequency/strand, A is the absorbance produced from a given amount of non-damaged DNA template, and Ad is the absorbance produced from a given amount of damaged DNA template induced by a particular dose of the ruthenium compound. Therefore, Ad/A gives the fraction of non-damaged template at a given dose [55].
RNA extraction and reverse transcription-polymerase chain reaction PCR (RT-PCR): The tested cells were seeded at a density of 106 cells/mL into 6-well culture plates. After 48 h of treatment with the IC50 of [Ru(p-cymene)(dppm)Cl2], cells were collected and total RNA was isolated using the RNeasy® Mini Kit (Qiagen, Germany) following the manufacturer’s instructions. Afterwards, the first strand complementary DNA (cDNA) synthesis was initiated using a 2-step RT-PCR kit (Vivantis, USA). RT-PCR was performed with the SYBR green-based quantitative PCR (qRT-PCR) detection system. RT-PCR was performed in a final volume of 25 μL, consisting of the ruthenated-cDNA template (100 ng), QuantiFast SYBR green PCR master mix (12.5 μL), and forward and reverse primers (0.5 μM of each). The gene-specific primer sequences were as follows: BRCA1: 5/-GCCAGTTGGTTGATTTCCACC-3/ (forward) and 5/-GTCAAATGTCTCCCCAAAAGC-3/ (reverse); p53: 5/-GGTCTCCCCAAGGCGCACTGG-3/ (forward) and 5/-AGGCTGGGGGCACAGCAGGCC-3/ (reverse); p21: 5/-GACACCACTGGAGGGTGACT-3/ (forward) and 5/-CAGGTCCACATGGTCTTCCT-3/ (reverse), and the sequence of β-actin: 5/-GGACTTCGAGCAAGAGATGG-3/ (forward) and 5/-AGCACTGTGTTGGCGTACAG-3/ (reverse) as an internal control. All samples were composed in triplicate and reacted in the following cycling condition: 1 cycle of an initial step at 95 °C for 5 min, followed by 35 cycles of 95 °C for 10 s, 60 °C for 30 s and 72 °C for 30 s. The melt curve analysis was performed by measuring fluorescence during the annealing step at a steadily increasing temperature from 55 to 90 °C on an ABI-Prism 7300 analytical thermal cycler (Applied Biosystems). The 2-ΔΔCT method was employed to analyze the RT-PCR data. β-actin mRNA expression was used as a normalization by subtracting the value of each gene of interest in each sample [56].
Protein extraction and western blotting analysis: After incubation for 48 h with the IC50 of [Ru(p-cymene)(dppm)Cl2] at 37 °C, cells were washed twice with PBS, harvested, and lysed for 5 min in wells containing 200 μL of lysis buffer (50 mM Tris (pH 8.0), 5 mM EDTA, 5 mM NaCl, 100 mM PMSF, 2% SDS, 1% Triton X-100,) placed on ice. The lysate proteins were isolated by centrifugation at 16,000 × g at 4 °C for 10 min. The concentration of the crude protein in each sample was determined using the Bradford assay. Protein samples of 50 μg were mixed with electrophoresis buffer and boiled for 10 min followed by separation by electrophoresis on a 6% SDS-polyacrylamide gel. Then, using a semi-dry electroblotter, separated proteins in the gel were transferred to a nitrocellulose membrane through a transfer buffer (14.4 g of glycine, 3.03 g of Tris base, 200 mL of methanol, and double distilled water to final volume of 1 liter) in a current of 400 mA for 6 h. The nitrocellulose membrane was incubated with shaking for 4 h in 10% bovine serum albumin (BSA) blocking reagent buffer in Tris-bufferedsaline (TBS). After removal of the blocking reagent buffer, the nitrocellulose membrane was further incubated overnight at 4 °C with an appropriate dilution in TBS buffer of primary antibody, anti-BRCA1 (Ab-1) mouse (MS110) antibody (Calbiochem (EMD Millipore)), mouse anti-p53 horseradish peroxidase-conjugated antibody (R&D Systems), and mouse monoclonal anti-p21 (R&D System). Then, the primary antibody was removed and washed 8 times for 5 min with a washing buffer of TBS with 0.1% Tween® 20 (TBST). The blot was incubated with shaking for 4 h with the secondary antibody, an HRP conjugated, goat anti-mouse IgG (HAF, R&D Systems) at a 1:5000 dilution in TBS with 10% BSA. A protein loading control, anti β-actin clone C4 (Calbiochem (EMD Millipore)), was diluted at a 1:5000 in TBS with 10% BSA. The blot was rinsed 8 times for 5 min with TBST, and incubated with shaking for 1 h with anti-mouse IgG horseradish peroxidase conjugated secondary antibody (R&D Systems) at a dilution of 1:1000 in TBS with 10% BSA. The blot was again rinsed with TBST. The immunoreactive signal detections were visualized by enhanced chemiluminescence using mixed equal volumes of stable peroxide solution and Lumino/Enhancer solution (Bio-Rad). The blot was immediately exposed to the working solution for 5 min. Finally, the blot was carefully placed in a film cassette, removing air bubbles before exposure to X-rays in darkness. The experiments were performed in triplicate.
Statistical analysis: SPSS version 22.0 was used for statistical analysis. Each value was expressed as a mean ± standard error of the mean (SEM) and comparisons between two groups were made by one-way ANOVA. Group differences resulting in * p < 0.01 were considered statistically significant.
[Ru(p-cymene)(dppm)Cl2] was highly cytotoxic toward human breast cancer cells: Cell growth inhibition by
[Ru(p-cymene)(dppm)Cl2] was initially evaluated by MTT assay. The ruthenium compound displayed a high degree of cytotoxicity against all three breast cancer cell lines (Table 1) but, within the IC50 values, no cytotoxic effect was exhibited on the growth of CCD841CoN normal human colon epithelial cells (data not shown). The inhibition of cell growth exhibited was dose-dependent (Figure 2a). Notably, 5 µM of [Ru(p-cymene)(dppm)Cl2] reduced the cell viability of all tested cells by more than 80%, which was only slightly less than the reduction produced by a concentration of 100 μM. The dppm ligand reduced cellular viability by more than 50% at a concentration higher than 25 µM. Triple-negative BRCA1-deficient HCC1937 cells were considerably more sensitive to the ruthenium compound (IC50 = 0.9 µM) than triple-negative BRCA1 wild-type MDA-MB-231 (IC50 = 1.8 µM) and sporadic BRCA1 wild-type MCF-7 cells (IC50 = 2.5 µM). The ruthenium compound displayed a greater cytotoxicity than its dppm ligand and cisplatin against all the cell lines (Table 1, Figure 2b). MCF-7, MDA-MB-231, and HCC1937 cells were respectively 14, 10, and 13 times more sensitive to the ruthenium compound than to the dppm ligand alone. Remarkably, the cytotoxicity of the ruthenium compound was greater than the cytotoxicity of cisplatin by a factor of 17, 71, and 26 times against MCF-7, MDA-MB-231, and HCC1937 cells, respectively.
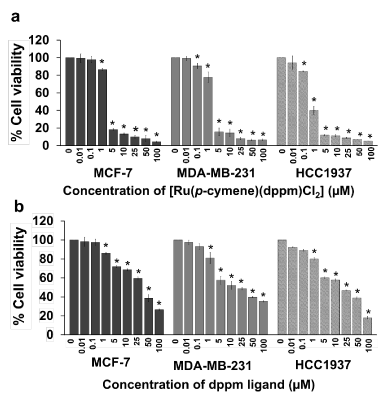
Figure 2. The cytotoxicity of [Ru(p-cymene)(dppm)Cl2] (a) and the dppm ligand (b) toward BRCA1-associated breast cancer cells was determined using the MTT assay. MCF-7, MDA-MB-231, and HCC1937 cells were exposed to various doses of the ruthenium compound and its ligand for 48 h in an incubator at 37 °C in 5% carbon dioxide (CO2). Each concentration was replicated at least four times. The experiments were performed at three separate times and the bar represents the standard error of the experiments. Statistically significant differences in comparison with the untreated control are denoted by *p < 0.01
Table 1. Comparative half-maximal inhibitory concentration (IC50) profiles (μM) of MCF-7, MDA-MB-231, and HCC1937 cells after 48 h treatment with the [Ru(p-cymene) (dppm)Cl2] complex and its dppm ligand alone. In addition, the IC50 values of cisplatin were included in parallel as a metal-based reference drug comparison. The mean values ± standard error of the mean (SEM) was collected as the average of three independent experiments. In the experiment, at least four parallel wells were set containing each concentration of the [Ru(p-cymene) (dppm)Cl2] complex and its ligand
A statistically significant difference was used to evaluate the IC50 values of the same compound based on cell lines and indicated by * p< 0.01, and to evaluate the IC50 values of the compounds based on a single cell line and indicated by ** p< 0.01
Differential profiles of the intracellular distribution of the ruthenium compound: The efficacy of anticancer activity normally depends on the cellular uptake of the drug. The total intracellular ruthenium levels were monitored by ICP-MS anlysis. The total percentage of the amount of ruthenium detected in the intracellular compartments of Ru-treated breast cancer cells after 48 h of ruthenium treatment was significantly different in the cytoplasm, mitochondria, and nucleus (Figure 3). The distribution of ruthenium atoms was noticeably different in each cell line. In MCF-7 cells, the ruthenium content was detected predominantly in the cytoplasm (61.9%), followed by the mitochondria (26.8%) and the nuclear fraction (11.7%). In MDA-MB-231 cells, the ruthenium atom mainly accumulated in the mitochondrial fraction (80.8%) with equal smaller amounts of ruthenium in the nuclear fraction and cytoplasm. In HCC1937 cells, the ruthenium atom was mainly detected in the nuclear fraction (92.0%) with a small amount in both the cytoplasm and nuclear fraction.
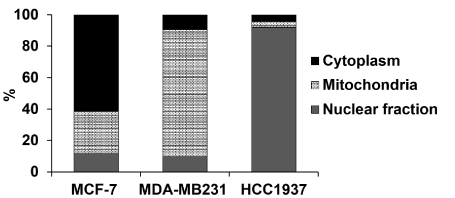
Figure 3. The chart shows the intracellular distribution of [Ru(p-cymene)(dppm)Cl2] in MCF-7, MDA-MB-231 and HCC1937 cells. Cells were incubated with the IC50 of the ruthenium compound (Table 1) at 37 °C for 48 h. The ICP-MS analytical technique was used to detect subcellular ruthenium atoms in the cytoplasm, nuclear, and mitochondria fractions. The percentage of ruthenium content present in each of the subcellular fractions was calculated from the sum of the mass of ruthenium atoms detected in all three fractions
Ruthenium compound induced cell cycle arrest and apoptotic cell death: The effect of the ruthenium compound at its IC50 value on cell cycle distribution was estimated by propidium iodide (PI) staining using flow cytometric analysis. The cell cycle arrest progression in all tested cell lines presented significant differences between treatment and control groups (Figure 4). The populations of Ru-treated MCF-7 and HCC1937 cells arrested at the G2/M phase (15.7% and 24.0%, respectively) were approximately twofold the populations of untreated control cells arrested at the same stage. By contrast, the Ru-treated cell populations at the G0/G1 and S phases were smaller than the untreated cell populations in the same phases (Figures 4a and 4c). However, in the MDA-MB-231 cells, the number of Ru-treated cells arrested in the G0/G1 phase was significantly higher than the number of untreated cells (77.7% compared to control 68.6%) (Figure 4b). Consequently, the accumulation of treated cells at the S and G2/M phase was lower than the number of untreated cells. The results indicated that the ruthenium compound displayed a specific difference in alteration of cell cycle progression.
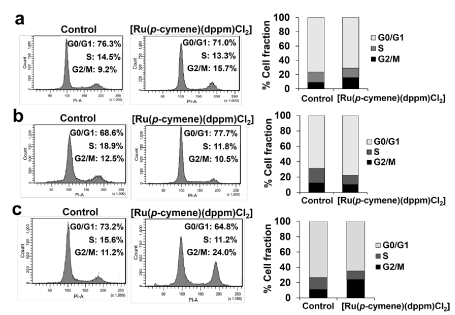
Figure 4. Cell cycle progression induced by [Ru(p-cymene)(dppm)Cl2] was analyzed using flow cytometry. MCF-7 (a), MDA-MB-231 (b) and HCC1937 (c) cells were exposed to the ruthenium compound at the IC50 for 48 h at 37 °C. Cells were then stained with propidium iodide (PI). The DNA content histograms show the majority phases of the cell cycle in the G0/G1, S, and G2/M (from the left to the right). The experiment was conducted in triplicate
In addition, the percentage of apoptotic cell death was determined by double-staining treated cells with Annexin V and PI. The induction of apoptotic cells was observed in relation to the total amount of early- and late stage-apoptotic cells. In the Ru-treated cells, a greater increase in the percentage of apoptotic cells was observed in MDA-MB-231 (70%) than in HCC1937 (51%) and MCF-7 (35%) cells (Figure 5).
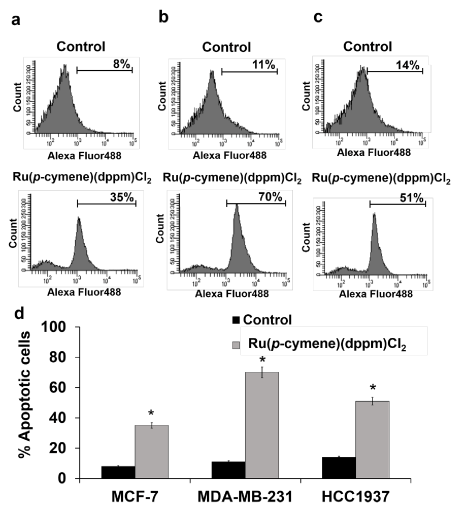
Figure 5. [Ru(p-cymene)(dppm)Cl2] induced apoptotic cell death in MCF-7 (a), MDA-MB-231 (b) and HCC1937 (c) cells. Cells were incubated for 48 h with the ruthenium compound at its IC50 at 37 °C. Flow cytometric profiles of Annexin-V FITC staining in a representative experiment are shown. The experiments were performed at three separate times and the bar represents the standard error of the experiment (d). Statistically significant differences in comparison with the untreated control are denoted by *p < 0.01
[Ru(p-cymene)(dppm)Cl2] significantly suppressed BRCA1 amplification in both BRCA1-deficient and -proficient triple-negative breast cancer cells: The effect of the ruthenium compound on cellular BRCA1 damage in MCF-7, MDA-MB-231, and HCC1937 cells was investigated using a quantitative PCR-based assay (QPCR). After 48 h of treatment with the ruthenium compound, BRCA1 amplification was reduced in all tested cells. The reduction was correlated with dose increments of the ruthenium compound (Figure 6a). This result implied that the ruthenium compound is capable of inhibiting the amplification of the BRCA1 gene in a dose-dependent manner. The degree of BRCA1 damage was significantly higher in HCC1937 cells than in MDA-MB-231 and MCF-7 cells at every concentration of the ruthenium compound. Notably, the percentage of BRCA1 amplification was reduced by half in HCC1937 and MDA-MB-231 cells at ruthenium doses of approximately 50 μM and 80 μM, respectively (Figure 6b), with approximately 0.5-1 ruthenium atom per BRCA1 fragment (Figure 6c). Meanwhile, a 500 μM concentration of the ruthenium compound only slightly impacted BRCA1 amplification in MCF-7 cells (<10%) (Figure 6b). The results indicated that the ruthenium compound was significantly more capable of blocking BRCA1 replication in HCC1937 and MDA-MB-231 cells than in MCF-7 cells.
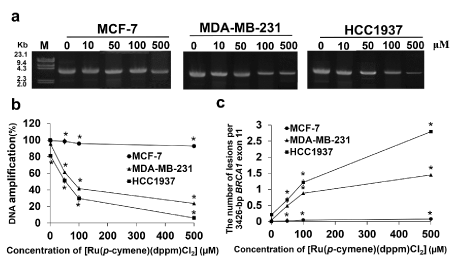
Figure 6. Cellular BRCA1 damage due to [Ru(p-cymene) (dppm)Cl2] in MCF-7, MDA-MB-231, and HCC1937 cells was investigated using QPCR. Cells were incubated for 48 h with various doses of the ruthenium compound at 37 °C before cellular genomic DNA was collected. The 3,426-bp BRCA1 exon 11 fragments was then amplified using specific forward and reverse primers in a PCR reaction. Amplified BRCA1 products were electrophoresed on 1% agarose gel. The gel was stained with ethidium bromide and illuminated under ultraviolet light (a). The percentage of DNA amplification induced by ruthenium compound (b), and the ruthenium compound induced the lesions per the 3,426-bp BRCA1 exon 11 fragment was calculated by the Poisson equation [54] and plotted with various concentrations of [Ru(p-cymene)(dppm)Cl2] (c). The experiments were carried out at three independent times. Statistically significant changes by comparison with the Ru-untreated control are denoted by *p < 0.01
[Ru(p-cymene)(dppm)Cl2] had distinct regulation the expression of BRCA1, p53 and p21 mRNAs: The mRNA expression of genes of interest induced by the ruthenium compound was determined using real-time quantitative RT-PCR. The ruthenium compound exhibited a different level of regulation on the expression of BRCA1, p53, and p21 mRNAs in all three cell lines (Figure 7). In MCF-7 cells, the expression of p21 and p53 mRNA was downregulated (Figure 7a), but in MDA-MB-231 cells (Figure 7b) the expression of p21 and p53 mRNA was upregulated. The expression of BRCA1 mRNA was suppressed in Ru-treated HCC1937 cells, while it was upregulated in Ru-treated MCF-7 cells and remained unchanged in Ru-treated MDA-MB-231 cells (p < 0.01).
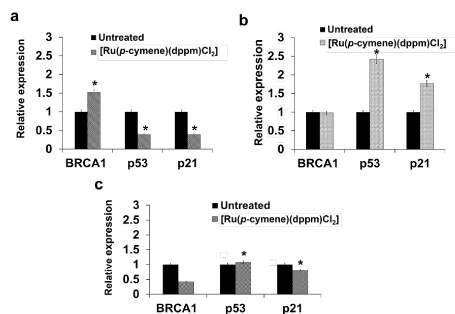
Figure 7. The charts show the effect of [Ru(p-cymene)(dppm)Cl2] treatment on mRNA expression of MCF-7 (a), MDA-MB-231 (b), and HCC1937 (c) cells. Cells were incubated for 48 h with [Ru(p-cymene)(dppm)Cl2] at the IC50 before total RNAs were extracted. The quantity of transcriptional of the genes was evaluated by a real-time quantitative RT-PCR. The 2-ΔΔCT method was employed to analyze the RT-PCR data. β-actin mRNA expression was used to normalize the value of each gene in each sample and relative to the expression of Ru-untreated control cells. The experiments were performed in three independent times. Statistically significant alteration in the mRNA expression of each gene in comparison with the untreated control are denoted by *p < 0.01
[Ru(p-cymene)(dppm)Cl2] downregulated BRCA1 but upregulated p53 and p21 protein expression: Whole-cell protein extracts of ruthenium-treated cells were immunoblotted using specific antibodies. The results revealed that reductions in the levels of the BRCA1 protein were similar in all Ru-treated cells (Figure 8a). Treatment with the ruthenium compound suppressed BRCA1 protein expression more in HCC1937 cells than in MDA-MB-231 and MCF-7 cells, with a 4-, 1.6- and 1.3-fold, respectively, compared with the Ru-untreated cells (Figure 8b). On the other hand, the expression of p53 and p21 proteins was upregulated. Upregulation of the relative expression of both genes was highest in MCF-7 > MDA-MB-231 > HCC1937.
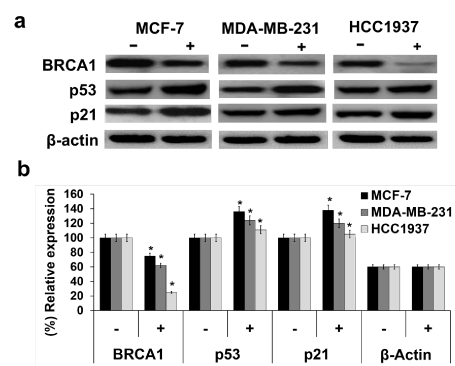
Figure 8. The effect of [Ru(p-cymene) (dppm)Cl2] on BRCA1, p53, and p21 protein expression was investigated in MCF-7, MDA-MB-231, and HCC1937 cells. Cells were exposed for 48 h to the ruthenium compound at its IC50 at 37 °C. The crude extracted proteins were separated on 6% SDS-PAGE, and the gel was transferred to a nitrocellulose membrane. The target proteins were detected by primary anti-BRCA1, anti-p53, and anti-p21 antibodies, membranes were further probed with secondary anti-mouse horseradish peroxidase conjugated antibody (a). The equality of protein loading in each sample was validated by an actin antibody. The band intensity of each blot was quantified using a Bio-Rad Molecular Imager with the Molecular Dynamics program (b). The experiments were carried out in triplicate. Statistically significant alterations in comparison with the Ru-untreated control are denoted by * p<0.01
We evaluated a ruthenium(II)-arene compound containing a 1,1-bis(diphenylphosphino)methane ligand, termed [Ru(p-cymene)(dppm)Cl2], for anticancer activity and BRCA1 inhibition against triple-negative BRCA1-deficient HCC1937, triple-negative BRCA1 wild-type MDA-MB-231, and BRCA1 wild-type MCF-7 breast cancer cells. In a dose-dependent manner, the ruthenium compound showed a higher cytotoxicity against all three breast cancer cells than the dppm ligand alone and the standard anticancer platinum-based drug cisplatin. The dppm ligand alone also exerted more cytotoxic effect than cisplatin. This class of ruthenium(II)-arene (diphenylphosphine) derivatives has shown good kinetic stability and reactivity due to chemical structures in which Ru(II) is surrounded mainly by coordinations with π-conjugated carbons in p-cymene for bonding, and the dppm ligand employs the lone electron pair of the phosphorus atom [57-59]. It was inferred that the chelating dppm ligand might have a strong effect on cell growth inhibition. It is likely that the chelating diphosphine ligand in dppm, with phosphorus serving as the electron donor atom and p-cymene forming intermolecular p-p stacking, provides an important lipophilicity to [Ru(p-cymene)(dppm)Cl2] that facilitates cellular uptake through the membrane, enhancing the anticancer activity [51,60]. It is most likely that the ability of the complex to permeate the cell membrane is enhanced by chelation. Previous studies have shown that ruthenium(II) complexes bearing phosphine and its derivative ligand exhibited promising anticancer activity against several cancer cell lines at a low IC50 [48,50,61-63]. The presence of three ligands tethering the ruthenium metal center can contribute to improved lipophilicity and cellular uptake, which facilitates permeation of the tumor cell membrane allowing interaction with molecularly targeted molecules.
Several lines of evidence have demonstrated that differences in cytotoxicity were closely associated with differential subcellular distributions of anticancer metal-based compounds, cell cycle progression, induction of apoptotic cell death, and alterations in gene expression profiles [46,64,65]. [Ru(p-cymene)(dppm)Cl2] was differently accumulated and had distinct regulatory effects on cell cycle arrest in all tested cells. In the MCF-7 and HCC1937 cells, significantly increased apoptotic cell numbers were observed at the G2/M phase but in the MDA-MB-231 cells, apoptotic cell numbers were high at the G0/G1 phase and a greater percentage of apoptotic cells was observed in the MDA-MB-231 cells than in the HCC1937 and MCF-7 cells. It has previously been demonstrated that NAMI-A and RAPTAs inhibited cell growth by triggering cell cycle arrest (and subsequently apoptotic cell death) at the G2/M phase [66,67]. Here, the greater sensitivity of the HCC1937 cells toward this ruthenium compound resulted in part from the greater accumulation of ruthenium atoms in the nuclear compartment. This event facilitated the formation of Ru-DNA adducts as well as Ru-BRCA1 adducts in HCC1937 and MDA-MB-231 cells. It might also be associated with a defect of BRCA1 functions (5382insC BRCA1 mutation) in the HCC1937 cells that prevented the repair of DNA damage produced by the ruthenium compound, which ultimately led to apoptotic cell death [68-70]. This finding is consistent with findings of our previous studies that treatment with ruthenium(II) polypyridyl derivatives and RAPTAs caused more damage to the cellular BRCA1 gene in triple-negative BRCA1-defective breast cancer HCC1937 cells than in the triple-negative BRCA1 wild-type breast cancer MDA-MB-231 and the sporadic BRCA1 proficient breast cancer MCF-7 cells [46,53,55].
The underlying molecular mechanisms by which the ruthenium compound induces apoptosis in HCC1937 cells produced a similar inhibition of DNA replication, transcription and translation, leading to apoptotic cell death induction [71]. Some researchers have recently found that ruthenium(II)-arene compounds have an impact on more diverse cellular molecular targets than DNA, suggesting a different biochemical mode of action compared to classical platinum anticancer drugs [53,72-75]. Cell proliferation was more effectively inhibited in the MDA-MB-231 cells than in the sporadic MCF-7 cells. This evidence could be due to the greater accumulation of ruthenium atoms in the mitochondria of the MDA-MB-231 cells. The interference with mitochondrial membrane potential released molecules that can activate the apoptotic pathways, ultimately triggering a significant increase in apoptotic cells [76]. It was previously observed that doxorubicin or docetaxel had a greater effect on the highly invasive MDA-MB-231 cells, compared with the non-invasive MCF-7 cells [77]. In addition, MDA-MB-231 cells lack the Bcl-2 apoptosis inhibitory protein that regulates mitochondria permeability [78,79]. Triple-negative BRCA1 competent cells might induce multiple pathways, leading to apoptotic cell death through upregulation of the epidermal growth factor (EGF)-induced nuclear factor κB (NF-κB) [67,80]. Moreover, emerging studies have shown that mitochondrial DNA (mtDNA) is a potential target for cancer therapy [81,82]. Therefore, it might be possible that the phenyl group in a chelating dppm ligand binds to mtDNA and induces mtDNA damage, hence interfering with the function of mtDNA by reducing the amplification and copy number of mtDNA and affecting the transcriptional level of mitochondria-encoded genes that might be responsible for the increased apoptosis in this cell line [81,82]. The induction of apoptotic cell death represents an important mechanism in the clinical evaluation of therapeutic potentials [47,83], implying an alternative mechanism for metal-based anticancer compounds [84].
The lower IC50 values for MCF-7 cells in this study could be correlated to the different intracellular distributions of ruthenium compound and significantly lower numbers of apoptotic cells, compared with MDA-MB-231 and HCC1937 cells. Indeed, MCF-7 cells lack the expression of caspase-3, which is an absolutely crucial effector protein for apoptosis induction [85]. Alternatively, this class of ruthenium(II)-arene (diphenylphosphino) derivatives might directly inhibit specific proteins involved in cellular intrinsic or signal transduction pathways, giving rise to anticancer activity in these breast cancer cells [86].
Several lines of evidence have demonstrated that cancerous cells with dysfunctional BRCA1 are defective in DNA double strand break (DSB) repair, transcriptional regulation, and protein ubiquitination [7,18,70]. Cancer cells carrying inactivated BRCA1 show increased sensitivity to chemotherapeutic drugs or DNA-damaging agents that ultimately lead to genomic instability and cell death. Therefore, cellular damage to the BRCA1 gene by anticancer ruthenium compounds could interfere with BRCA1 multi-functions, leading to cancer cell death. To address this question, we investigated whether [Ru(p-cymene)(dppm)Cl2] affected the cellular BRCA1 gene. The ruthenium compound at its IC50 value caused a significant reduction in BRCA1 amplification in HCC1937 and MDA-MB-231 cells while it caused only slight reduction in MCF-7 cells. This may partly be due to the different cellular responses to ruthenium treatment exhibited by each molecular subtype of breast cancer cells [2,3,87]. The present data agreed very well with our previous studies of the inhibition of BRCA1 amplification induced by cisplatin, carboplatin, RAPTAs, and metallo-intercalator ruthenium(II) polypyridyl compounds [46,53-55,88,89]. It is likely that the drug exerted its inhibitory effect through distortion of DNA base pairs that led to the intervention of the BRCA1 polymerization process [54,89]. Importantly, with regard to the inhibition of BRCA1 replication, both triple-negative cells used in this study, BRCA1-mutated HCC1937 and BRCA1-wild-type MDA-MB-231, are significantly more susceptible to the ruthenium(II)-arene (dppm) compound than the sporadic BRCA1-competent MCF-7 cells. This ruthenium compound exerts its inhibitory effect on DNA replication of the BRCA1 gene in a similar manner in both MDA-MB-231 and HCC1937 cells. The higher level of inhibition of BRCA1 amplification in MDA-MB-231 and HCC1937 cells is most likely linked to the accumulation of ruthenium atoms in the nuclear and mitochondrial compartments, respectively, of these cells. It is, therefore, easier for the ruthenium compound to damage the BRCA1 gene, DNA replication apparatus and relevant signal transduction pathways involved in cell proliferation, cell cycle progression and programmed cell death. In addition, the RT-PCR data revealed that the ruthenium compound differently regulated the expression of BRCA1, p53 and p21 mRNAs in ways that were disproportional to their respective proteins (Figures 7 and 8). Increased breast cancer cell death was associated with an increased expression of p53 and p21 proteins in MCF-7, MDA-MB-231, and HCC1937 cells in the order shown schematically in figure 8.
Globally, the TNBC molecular subtype is known to offer a poor prognosis and outcome. Presently, no recognized standard regimen or therapy is available. BRCA1 expression limits the efficacy of treatment for TNBC breast cancer patients. Preclinical and clinical studies showed that TNBCs that expressed BRCA1 were more resistant to platinum drugs than TNBCs that did not [18,25,90,91] and therefore, the expression level of BRCA1 has been referred as a molecular biomarker to predict the treatment response among breast cancer patients [22-25]. In the present study, the expression level of the BRCA1 protein was significantly reduced in all three breast cancer cell lines after exposure to [Ru(p-cymene)(dppm)Cl2]. Expression level reductions were greatest in HCC1937, followed by MDA-MB-231, and lowest in MCF-7. The data are in consensus with our previous studies that found the expression of the BRCA1 protein in these cells was reduced after treatment with ruthenium(II) polypyridyl compounds [55] and RAPTAs [46,53]. In addition, the expression of BRCA1 modulated the chemosensitivity of BRCA1-mutated HCC1937 cells [8,18]. Together, the present findings provide insights into the underlying molecular mechanism of action of the anticancer activity of [Ru(p-cymene)(dppm)Cl2] against BRCA1-associated breast cancer cells, and increase the possibility of targeting BRCA1 in chemotherapeutic treatment of aggressive triple-negative BRCA1-associated breast cancers.
The data demonstrated that the ruthenium complex, [Ru(p-cymene)(dppm)Cl2], induced a differential cellular response in breast cancer cells. The ruthenium compound revealed a high degree of cytotoxicity against three breast cancer cells. Triple-negative BRCA1 deficient HCC1937 cells were significantly more sensitive to the ruthenium compound than triple-negative BRCA1 wild-type MDA-MB-231 and sporadic BRCA1 wild-type MCF-7 cells. Treatment with the ruthenium compound displayed much more effective against all selected cell lines than the dppm ligand alone and cisplatin. The cytotoxic effect of the ruthenium compound was correlated with the pattern of its accumulation inside the cells, and the resulting changes in cell cycle progression and apoptosis induction. Increased breast cancer cell death was associated with an enhanced expression of p53 and p21 mRNAs and their respective proteins. Damage of the BRCA1 gene induced by the ruthenium compound was found to be much higher in HCC1937 cells than MDA-MB-231 and MCF-7 cells. The ruthenium compound differently regulated the expression of BRCA1 mRNA in these cell lines. The expression of the BRCA1 protein was reduced the most in HCC1937 and the least in MCF-7 cells. These findings could provide a meaningful evidence to support the development of ruthenium-based compounds for effective, targeted therapy of breast cancers. It is expected that the obtained data will be beneficial for future therapeutic strategies that employ selective, molecular targeting of BRCA1 by metal-based anticancer agents in the treatment of BRCA1-associated and triple-negative breast cancer subtypes.
This research was funded by the National Research Council of Thailand and Prince of Songkla University, (PHA610093S, and PHA590396S), the Graduate School, Prince of Songkla University (PHA6202079S), and the Prince of Songkla University Scholarship for Ph.D. Program (to T.N., PSU 019/2557). N.L. acknowledges financial support from the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Ministry of Higher Education, Science, Research and Innovation and the Faculty of Science.
Conceptualization and design, A.R., T.N., and N.L.; methodology, A.R., and T.N.; acquisition of data and analysis, A.R., and T.N.; writing and editing, T.N. and A.R.; resources, A.R.; supervision, A.R., and N.L.; project administration, A.R.; funding acquisition, A.R. All authors have read and agreed to the published revision of the manuscript.
We are grateful to Thomas Duncan Coyne for assistance with the English text, and the Pharmaceutical Laboratory Service Center, Faculty of Pharmaceutical Sciences, Prince of Songkla University for research facilities.
The authors declare that they have no conflicts of interest.
- Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70: 7-30.
- Perou CM, Sørlie T, Eisen MB, van de Rijn, M, Jeffreyk SS, et al. (2000) Molecular portraits of human breast tumours. Nature 406: 747-752. [Crossref]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, et al. (2001) Gene expression patterns breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98: 10869-10874. [Crossref]
- Ruijter de TC, Veeck J, Hoon de JP, Engeland van M, Tjan-Heijnen VC (2011) Characteristics of triple-negative breast cancer. J Cancer Res Clin Oncol 137: 183-192. [Crossref]
- Yao H, He G, Yan S, Chen C, Song L, et al. (2017) Triple-negative breast cancer: is there a treatment on the horizon? Oncotarget 8: 1913-1924.
- Yersal O, Barutca S (2014) Biological subtypes of breast cancer:Prognostic and therapeutic implications. World J Clin Oncol 5: 412-424.
- Ratanaphan A (2012) A DNA repair BRCA1 estrogen receptor and targeted therapy in breast cancer. Int J Mol Sci 13: 14898-14916.
- Hongthong K, Ratanaphan A (2016) BRCA1-associated triple-negative breast cancer and potential treatment for ruthenium-based compounds. Current Cancer Drug Targets 16: 606-617. [Crossref]
- Garrido-Castro AC, Lin NU, Polyak K (2019) Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov 9: 176-198.
- Wu J, Mamidi TKK, Zhang L, Hicks C (2019) Integrating germline and somatic mutation information for the discovery of biomarkers in triple-negative breast cancer. Int J Environ Res Public Health 16: 1055. [Crossref]
- Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, et al. (2018) A randomised phase III trial of carboplatin compared with docetaxel in BRCA1/2 mutated and pre-specified triple negative breast cancer “BRCAness” subgroups: the TNT Trial. Nat Med 24: 628-637.
- Caramelo O, Silva C, Caramelo F, Frutuoso C, Almeida-Santos T, et al. (2019) The effect of neoadjuvant platinum-based chemotherapy in BRCA mutated triple negative breast cancers-systematic review and meta-analysis. Hered Cancer Clin Pract 17: 11.
- McAndrew N, DeMichele A (2018) Neoadjuvant chemotherapy considerations in triple-negative breast cancer. J Target Ther Cancer 7: 52-59. [Crossref]
- Poggio F, Bruzzone M, Ceppi M, Ponde NF, Valle GL, et al. (2018) Platinum-based neoadjuvant chemotherapy in triple-negative breast cancer: a systematic review and meta-analysis. Ann Oncol 29: 1497-1508.
- Walsh EM, Shalaby A, O'Loughlin M, Keane N, Webber MJ, et al. (2019) Outcome for triple negative breast cancer in a retrospective cohort with an emphasis on response to platinum-based neoadjuvant therapy. Breast Cancer Res Treat 174: 1-13.
- Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363: 1938-1948.
- Anders CK, Carey LA (2009) Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer 9: S73-S81. [Crossref]
- Tassone P, Tagliaferri P, Perricelli A, Blotta S, Quaresima B, et al. (2003) BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br J Cancer 88: 1285-1291.
- Husain A, He G, Venkatraman ES, Spriggs DR (1998) BRCA1 upregulation is associated with repair-mediated resistance to cisdiaminedichloroplatinum (II). Cancer Res 58: 1120-1123.
- Quinn JE, Kennedy RE, Mullan PB, Gilmore PM, Carty M, et al. (2003) BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Res 63: 6221-6228. [Crossref]
- Quinn JE, James, CR, Stewart GE, Mulligan JM, White P, et al. (2017) BRCA1 mRNA expression levels predict for overall survival in ovarian cancer after chemotherapy. Clin Cancer Res 13: 7413-7420.
- Toss A, Cristofanilli M (2015) Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res 17: 60.
- Prat A, Carey LA, Adamo B, Vidal M, Tabernero J, et al. (2014) Molecular features and survival outcomes of the intrinsic subtypes within HER2-positive breast cancer. J Natl Cancer Inst 106: dju152.
- Moy I, Lin Z, Rademaker AW, Reierstad S, Khan SA, et al. (2013) Expression of estrogen-related gene markers in breast cancer tissue predicts aromatase inhibitor responsiveness. PLoS One 8: e77543.
- Romagnolo APG, Romagnolo DF, Selmin OI (2015) BRCA1 as target for breast cancer prevention and therapy. Anticancer Agents Med Chem 15: 4-14
- Mylavarapu S, Das A, Roy M (2018) Role of BRCA mutations in the modulation of response to platinum therapy. Front Oncol 8: 16.
- Agrawal LS, Mayer IA (2014) Platinum agents in the treatment of early-stage triple-negative breast cancer: is it time to change practice? Clin Adv Hematol Oncol 12: 654-658. [Crossref]
- Kelland L (2007) The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 7: 573-584.
- Rabik CA, Dolan ME (2007) Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev 33: 9-23.
- Dilruba S, Kalayda GV (2016) Platinum-based drugs: past, present and future. Cancer Chemother Pharmacol 77: 1103-1124.
- Oun R, Moussa YE, Wheate NJ (2018) The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans 47: 6645-6653.
- Ashworth A (2008) Drug resistance caused by reversion mutation. Cancer Res 68: 10021-10023.
- Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, et al. (2008) Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res 68: 2581-2586. [Crossref]
- Allardyce CS, Dyson PJ (2001) Ruthenium in medicine: current clinical uses and future prospects. Platinum Metals Rev 45: 62-69.
- Allardyce CS, Dyson PJ (2016) Metal-based drugs that break the rules. Dalton Trans 45: 3201-3209.
- Webb MI, Walsby CJ (2015) Albumin binding and ligand-exchange processes of the Ru (III) anticancer agent NAMI-A and its bis-DMSO analogue determined by ENDOR spectroscopy. Dalton Trans 44: 17482-17493.
- Zeng L, Gupta P, Chen Y, Wang E, Ji L, et al. (2017) The development of anticancer ruthenium(ii) complexes:from single molecule compounds to nanomaterials. Chem Soc Rev 46: 5771-5804.
- Ang WH, Dyson PJ (2006) Classical and non-classical ruthenium-based anticancer drugs: towards targeted chemotherapy. Eur J Inorg Chem 20: 4003-4018.
- Habtemariam A, Melchart M, Fernandez R, Parsons S, Oswald IDH, et al. (2006) Structure-activity relationships for cytotoxic ruthenium(II) arene complexes containing N, N-, N, O-, and O,O-chelating ligands. J Med Chem 49(23): 6858-6868. [Crossref]
- Wang F, Habtemariam A, van der Geer EPL, Fernandez R, Melchart M, et al. 2005. Controlling ligand substitution reactions of organometallic complexes:tuning cancer cell cytotoxicity. Proc Natl Acad Sci USA 102: 18269-18274.
- Beckford FA, Leblanc G, Thessing J, Shaloski M, Frost BJ, et al. (2009) Organometallic ruthenium complexes with thiosemicarbazone ligands: Synthesis, structure and cytotoxicity of [(eta-p-cymene) Ru(NS)Cl] (NS = 9-anthraldehyde thiosemicarbazones). Inorg Chem Commun 12: 1094-1098.
- Mokesch S, Schwarz D, Hejl M, Klose MHM, Roller A, et al. (2019) Fine-tuning the activation mode of an 1,3-indandione-based ruthenium(II)-cymene half-sandwich complex by variation of its leaving group. Molecules 24: 2373. [Crossref]
- Ude Z, Romero-Canelón I, Twamley B, Hughes DF, Sadler PJ, et al. (2016) A novel dual-functioning ruthenium(II)-arene complex of an anti-microbial ciprofloxacin derivative - anti-proliferative and anti-microbial activity. J Inorg Biochem 160: 210-217.
- Carsten AV, Anna KR, Rosario S, Lucienne JJ, Paul JD (2008) Influence of the diketonato ligand on the cytotoxicities of [Ru(η6‐p‐cymene) (R2acac) (PTA)]+ complexes (PTA = 1,3,5‐triaza‐7‐phosphaadamantane). Eur J Inorg Chem 10: 1661-1671.
- Muralisankar M, Dheepika R, Haribabu J, Balachandran C, Aoki S, et al. (2019) Design, synthesis, DNA/HSA binding, and cytotoxic activity of half-sandwich Ru(II)-arene complexes containing triarylamine-thiosemicarbazone hybrids. ACS Omega 4: 11712-11723. [Crossref]
- Ratanaphan A, Nhukeaw T, Hongthong K, Dyson PJ (2017) Differential cytotoxicity, cellular uptake, apoptosis and inhibition of BRCA1 expression of BRCA1-defective and sporadic breast cancer cells induced by an anticancer ruthenium(II)-arene compound, RAPTA-EA1. Anticancer Agents Med Chem 17: 212-220.
- Xu X, Lai Y, Hua ZC (2019) Apoptosis and apoptotic body: disease message and therapeutic target potentials. Biosci Rep 39: BSR20180992.
- Rodriguez-Barzano A, Lord RM, Basri AM, Phillips RM, Blacker AJ, et al. (2015) Synthesis and anticancer activity evaluation of η(5)-C5(CH3)4R ruthenium complexes bearing chelating diphosphine ligands. Dalton Trans 44: 3265-3270.
- Berners-Price SJ, Mirabelli CK, Johnson RK, Mattern MR, McCabe FL, et al. (1986) In vivo antitumor activity and in vitro cytotoxic properties of bis[1,2bis(diphenylphosphino)ethane] gold(I) chloride. Cancer Res 46: 5486-5493.
- Das S, Sinha S, Britto R, Somasundaram K, Samuelson AG, et al. (2010) Cytotoxicity of half sandwich ruthenium(II) complexes with strong hydrogen bond acceptor ligands and their mechanism of action. J Inorg Biochem 104: 93-104. [Crossref]
- Chuklin P, Chalermpanaphan V, Nhukeaw T, Saithong S, Chainok K, et al. (2017) Synthesis, X-ray structure of organometallic ruthenium (II) p-cymene complexes based on P- and N- donor ligands and their in vitro antibacterial and anticancer studies. J Organometallic Chem 846: 242-250.
- Koch RB (1969) Fractionation of olfactory tissue homogenates. Isolation of a concentrated plasma membrane fraction. J Neurochem 16: 145-157.
- Nhukeaw T, Hongthong K, Dyson PJ, Ratanaphan A (2019) Cellular responses of BRCA1-defective HCC1937 breast cancer cells induced by the antimetastasis ruthenium(II) arene compound RAPTA-T. Apoptosis 24: 612-622.
- Ratanaphan A, Canyuk B, Wasiksiri S, Mahasawat P (2005) In vitro platination of human breast cancer suppressor gene1 (BRCA1) by the anticancer drug carboplatin. Biochim Biophys Acta 1725: 145-151.
- Nhukeaw T, Temboot P, Hansongnern K, Ratanaphan A (2014) Cellular responses of BRCA1-defective and triple-negative breast cancer cells and in vitro BRCA1 interactions induced by metallo-intercalator ruthenium(II) complexes containing chloro-substituted phenylazopyridine. BMC Cancer 14: 73.
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Method 25: 402-408. [Crossref]
- Medicia S, Peanaa M, Nurchi VM, Lachowiczb JI, Crisponib G, et al. (2015) Noble metals in medicine: Latest advances. Coord Chem Rev 284: 329-350.
- Romero-Canelón I, Sadler PJ (2013) Next-generation metal anticancer complexes: multitargeting via redox modulation. Inorg Chem 52: 12276-12291.
- Zaki M, Hairatb S, Aazama ES (2019) Scope of organometallic compounds based on transition metal-arene systems as anticancer agents: starting from the classical paradigm to targeting multiple strategies. RSC Adv 9: 3239-3278.
- Yousefi R, Aghevlian S, Mokhtari F, Samouei H, Rashidi M, et al. (2012) The anticancer activity and hsa binding properties of the structurally related platinum(II) complexes. Appl Biochem Biotechno 167: 861-872. [Crossref]
- Berners-Price SJ, Norman RE, Sadler PJ (1987) The autoxidation and proton dissociation constants of tertiary diphosphines: relevance to biological activity. J Inorg Biochem 31: 197-209. [Crossref]
- Lopesa JCS, Damascenoc JL, Oliveirac PF, Guedesd APM, Tavaresc DC, et al. (2015) Ruthenium(II) complexes containing anti-inflammatory drugs as ligands: synthesis, characterization and in vitro cytotoxicity activities on cancer cell lines. J Braz Chem Soc 26: 1838-1847.
- Massai L, Fernández-Gallardo J, Guerri A, Arcangeli A, Pillozzi S, et al. (2015). Synthesis and characterisation of new chimeric ruthenium(II)-gold(I) complexes as improved cytotoxic agents. Dalton Trans 44: 11067-11076.
- Carminati PO, Mello SS, Fachin AL, Junta CM, Sandrin-Garcia P, et al. (2010) Alterations in gene expression profiles correlated with cisplatin cytotoxicity in the glioma U343 cell line. Genet Mol Biol 33: 159-168.
- Vekris A, Meynard D, Haaz MC, Bayssas M, Bonnet J, et al. (2004) Molecular determinants of the cytotoxicity of platinum compounds: the contribution of in silico research. Cancer Res 64: 356-362.
- Bergamo A, Gagliardi R, Scarcia V, Furlani A, Alessio E, et al. (1999) In vitro cell cycle arrest, in vivo action on solid metastasizing tumors, and host toxicity of the antimetastatic drug NAMI-A and cisplatin. J Pharmacol Exp Ther 289: 559-564. [Crossref]
- Chatterjee S, Biondi, I, Dyson, PJ, Bhattacharyya A (2011) A bifunctional organometallic ruthenium drug with multiple modes of inducing apoptosis. J Biol Inorg Chem 16: 715-724.
- Alli E, Sharma VB, Hartman AR, Lin PS, McPherson L, et al. (2011) Enhanced sensitivity to cisplatin and gemcitabine in Brca1-deficient murine mammary epithelial cells. BMC Pharmacol 11: 7.
- Norbury CJ, Zhivotovsky B (2004) DNA damage-induced apoptosis. Oncogene 23: 2797-2808.
- Tassone P, Di Martino MTD, Ventura M, Pietragalla A, Cucinotto I, et al. (2009) Loss of BRCA1 function increases the antitumor activity of cisplatin against human breast cancer xenografts in vivo. Cancer Biol Ther 8: 648-653.
- Brabec V, Novakova O (2006) DNA binding mode of ruthenium complexes and relationship to tumor cell toxicity. Drug Resistance Updates 9: 111-122.
- Adhireksan Z, Davey GE, Campomanes P, Groessl M, Clavel CM, et al. (2014) Ligand substitutions between ruthenium-cymene compounds can control protein versus DNA targeting and anticancer activity. Nat Commun 5: 3462-3475. [Crossref]
- Babak MV, Meier SM, Huber KVM, Reynisson J, Legin AA, et al. (2015) Target profiling of an antimetastatic RAPTA agent by chemical proteomics: relevance to the mode of action. Chem Sci 6: 2449-2456.
- Chotard F, Dondaine L, Balan C, Bettaieb A, Paul C, et al. (2019) Highly antiproliferative neutral Ru(II)-arene phosphine complexes. New J Chem 42: 8105-8112.
- Lee RFS, Chernobrovkin A, Rutishauser D, Allardyce CS, Hacker D, et al. (2017) Expression proteomics study to determine metallodrug targets and optimal drug combinations. Sci Rep 7: 1590.
- Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281: 1309-1312. [Crossref]
- Taherian A, Mazoochi T (2012) Different expression of extracellular signal-regulated kinases (ERK) 1/2 and phospho-Erk proteins in MBA-MB-231 and MCF-7 cells after chemotherapy with doxorubicin or docetaxel. Iran J Basic Med Sci 15: 669-677.
- Arif JM, Kunhi M, Bekhit AA, Subramanian MP, Al-Hussein K, et al. (2006) Evaluation of apoptosis-induction by newly synthesized phthalazine derivatives in breast cancer cell lines. Asian Pac J Cancer Prev 7: 249-252.
- Srivastava RK, Srivastava AR, Korsmeyer SJ, Nesterova M, Cho-Chung YS, et al. (1998) Involvement of microtubules in the regulation of Bcl2 phosphorylation and apoptosis through cyclic AMP-dependent protein kinases. Mol Cell Biol 18: 3509-3517.
- Biswas DK, Cruz AP, Gansberger E, Pardee AB (2000) Epidermal growth factorinduced nuclear factor κB activation: A major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci USA 97: 8542-8547. [Crossref]
- Burke CS, Byrne A, Keyes TE (2018) Highly selective mitochondrial targeting by a ruthenium(II) peptide conjugate: imaging and photoinduced damage of mitochondrial DNA. Angew Chem Int Ed Engl 57: 12420-12424.
- Wen S, Zhu D, Huang P (2013) Targeting cancer cell mitochondria as a therapeutic approach. Future Med Chem 5: 53-67.
- Poon IKH, Lucas CD, Rossi AG, Ravichandran KS (2014) Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 14: 166-180.
- Cao JJ, Zheng Y, Wu XW, Tan CP, Chen MH, et al. (2019) Anticancer cyclometalated iridium(III) complexes with planar ligands: mitochondrial DNA damage and metabolism disturbance. J Med Chem 62: 3311-3322.
- Jänicke RU (2009) MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res Treat 117: 219-221.
- Bergamo A, Masi A, Dyson PJ, Sava G (2008) Modulation of the metastatic progression of breast cancer with an organometallic ruthenium compound. Int J Oncol 33: 1281-1289. [Crossref]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, et al. (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10: 515-527.
- Chakree K, Ovatlarnporn C, Dyson PJ, Ratanaphan A (2012) Altered DNA binding and amplification of human breast cancer suppressor gene BRCA1 induced by a novel antitumor compound, [Ru(η6-p-phenylethacrynate)Cl2(pta)]. Int J Mol Sci 13: 13183-13202.
- Ratanaphan A, Temboot P, Dyson PJ (2010) In vitro ruthenation of human breast cancer suppressor gene 1 (BRCA1) by the antimetastasis compound RAPTA-C and its analogue carboRAPTA-C. Chem Biodivers 7: 1290-1302.
- Byrski T, Huzarski T, Dent R, Gronwald J, Zuziak D, et al. (2009) Response to neoadjuvant therapy with cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat 115: 359-363. [Crossref]
- Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, et al. (2010) Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J Clin Oncol 28: 1145-1153. [Crossref]