Bacterial cell wall synthesis pathway “Peptidoglycan Synthesis” is implicated in the development of antibiotics against various pathogens and provides an interesting target for therapeutic intervention. A medicinally valuable flavonoid “Rutin”, have been shown to be effective against various pathogenic bacteria. Hence in this study, the prospective role of Rutin interaction in the capability against peptidoglycan synthesizing enzymes has been investigated, where the binding pattern of rutin with Mur ligases, MraY, FemX, FemA, FemB, and PBP2 have been studied. However, among them, Rutin interacted better with PBP2, which catalyze two important steps (transglycosylation and transpeptidation) of cell wall synthesis using TGase and TPase domains respectively. The interaction pattern and stability of Rutin towards both domains were studied individually through docking and simulation studies, which suggested that Rutin affinity towards TGase domain was more and similar to the reference molecule Moenomycin, compared to the TPase domain. Thus, were reported herein, Rutin as a novel structural class for inhibitor development based on the results which provided better insights on the interactional studies against S. aureus by targeting TGase domain of PBP2.
cell wall, peptidoglycan synthesis, PBP2, S. aureus, rutin
The Staphylococcal cell wall is complex, dynamic, multilayered structures that play a variety of protective and adaptive roles for cell viability by providing the rigid exoskeleton which is essential for stabilizing the cell membrane against high internal osmotic pressures [1]. The major conserved component of the cell wall is peptidoglycan, a giant macromolecule consisting of linear glycan chain interlinked by short peptides [2]. Synthesis of peptidoglycan units is a multistep enzymatic process which can be divided into four phases (Figure 1) [3]; The first phase of peptidoglycan synthesis begins in the cytoplasm of the bacterial cell with the synthesis of UDP-Mur NAc pentapeptide, catalyzed by the cytoplasmic enzymes Mur ligases (MurA, B, C, D, E, F, G- which are functionally different from each other) [4,5]. The first committed step catalyzed by MurA enzyme with the transfer of enolpyruvyl moiety from phosphoenolpyruvate (PEP) to uridinediphosphate (UDP)-N-acetylglucosamine (GlcNAc) produces enolpyruvyl UDP-GlcNAc, which is catalysed by MurB through a NADPH-dependent conversion of UDP-GlcNAcEP to UDP-N-acetylmuramic acid. Further, MurC, MurD, MurE, and MurF catalyze the addition of L-Ala, D-Glu, L-Lys and D-Ala-D-Ala respectively onto the product of MurB (UDP-Mur NAc) [6]. Second phase includes membrane-associated steps, where formation of lipid I and lipid II are initiated by MraY [7] and MurG [8]. MraY catalyzes the formation of lipid I by transferring the MurNac-pentapeptide and GlcNAc to a membrane-bound acceptor. While, Lipid II formation is catalyzed by MurG by adding UDP-GlcNAc to lipid I and formed membrane bound disaccharide pentapeptide. Further in the third phase, disaccharide pentapeptide is modified by attaching the sequential addition of pentaglycine chain to the lysine residue of the pentapeptide by three homologous peptidyl transferases namely FemX/FmhB [9], FemA [10], and FemB [11]. Finally, in fourth phase, membrane bound disaccharide pentapeptide residue is transferred to the outside of the cell membrane to form linear peptidoglycans via transglycosylase and transpeptidase process, catalyzed by penicillin binding protein (PBP).
Based on the essentiality of cell wall for bacterial cell integrity, β-lactam antibiotics have been successfully targeted to inhibit the cell wall synthesis. However, these drugs besides producing enormous side effects have also made the organisms resistant to them. Since, natural compounds were reported to have pharmacological or biological activity that can be of therapeutic benefit in treating resistant strains, the present study was focused with the aim to study the interaction potentials of one of the flavonoid compound “Rutin” with other cell wall synthesizing enzyme through structure based molecular docking and simulation studies.
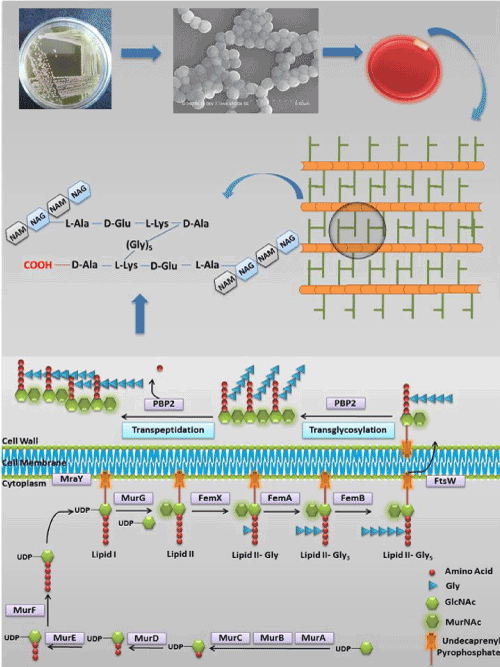
Figure 1. Schematic representation of the S. aureus peptidoglycan synthesis pathway
Homology modeling and structure refinement
Although eleven enzymes including Mur ligases, MraY, FemX, FemA, FemB, and PBP2 enzymes are found to be involved in the synthesis of peptidoglycan in S. aureus, among them crystal structure were revealed to be present for only four enzymes; MurB (PDB 1HSK), MurE (PDB 4C13), FemA (PDB 1LRZ), and PBP2 (PDB 2OLV) [12,13], necessitated the tertiary structures for other proteins (Mur A, C, D, F, G, MraY, Fem X, Fem B) to be modeled. Therefore, using Modeller 9.15 which included three steps as template selection, target-template alignment and model building, the models were predicted by retrieving the amino acid sequence of those proteins from the UniProt database and subjecting them to BLASTp search against the PDB database. Templates were selected for each of the protein on the basis of structural hits and retrieved from the Protein Data Bank (Table 1). Thereafter, 3-D model was generated, and best model was selected based on the lowest discrete optimized protein energy (DOPE) value [15] and further validated using SAVES (Structure Analysis and Verification Server) server [16, 17] (Table 2).
Table 1: Details of the templates used for modeling the structures of the Proteins
|
Proteins |
Protein UniProt ID |
Protein Length (Numbers of amino acids) |
Template (PDB ID) |
Query Coverage (%) |
|
MurA |
P84058 |
421 |
3SG1 |
99 |
|
MurC |
O31211 |
437 |
1GQQ |
96 |
|
MurD |
P0A091 |
449 |
3LK7A |
97 |
|
MurF |
K7XML5 |
452 |
3ZM5 |
95 |
|
MraY |
P0C1R8 |
321 |
4J72 |
94 |
|
MurG |
A5ISU9 |
356 |
1F0K |
91 |
|
FemB |
Q2FYR1 |
419 |
1LRZ |
99 |
|
FmhB |
Q2FVZ4 |
421 |
1LRZ |
96 |
Table 2. Validation of the modeled proteins
Proteins | ERRAT Score | VERIFY3D (%) | RMSD between template & modeled structure (Ǻ) | Residues in allowed region | DOPE value |
MurA | 83.495 | 97.86 | 0.099 | 97.60% | -49255.6 |
MurC | 70.163 | 88.98 | 0.776 | 94.90% | -46056.9 |
MurD | 79.365 | 91.09 | 0.123 | 96.20% | -52871.2 |
MurF | 70.045 | 89.16 | 0.263 | 97.00% | -46772.6 |
MraY | 96.141 | 95.01 | 0.161 | 95.00% | -44797.5 |
MurG | 93.966 | 82.02 | 0.544 | 97.20% | -47435 |
FemB | 74.572 | 82.83 | 0.366 | 96.60% | -48329.2 |
FmhB | 97.304 | 98.1 | 0.494 | 95.00% | -40185.6 |
MD simulation study
All generated models were simulated using GROMOS96 43a1 force field for 5000 ps for each of the modeled proteins and their energies were minimized using the steepest descent approach in the GROMACS 4.5.5 package and equilibrated (NVT and NPT) for 1000 ps to perform the final MD. The lowest potential energy (PE) conformations were selected from the simulation trajectory by plotting the Root Mean Square deviation (RMSD) graph and used for further analysis.
Molecular docking study
All cell wall synthesizing proteins were prepared for docking using Schrodinger protein preparation wizard, where proteins with the crystal structure (Mur B, E, FemA, and PBP2) were preprocessed and prepared by removing the water and other hetero- molecules (OPLS2005 force field) [18]. Grid for ligand docking was generated around the active site for each protein. Since, PBP2 catalyses transglycosylation and transpeptidation by TGase and TPase domain respectively, both of these domains were considered for the docking studies. Moreover, active site residues for all proteins were selected with the help of literature survey (Table 3). Finally, the prepared ligand-Rutin (ZINC053683228) was docked to each proteins using Glide package [19] available in Schrodinger software and analyzed manually. On the basis of docking results, best receptor-ligand complex was selected for MD simulation studies.
Table 3. Structure of proteins involved in peptidoglycan synthesis
S. No | Protein Name | 3-D Structure of Modeled Protein | Active Site Residues | References |
1 | MurA | 
| Leu113, Pro114, Gly115, Gly116,Cys117, Ser118, Iso119, Gly120, Ala121,
Arg122, Pro123 | [21] |
2 | MurB* |  > >
| Asp71, Tyr175, Arg176, Arg213, Ser226, Arg259,
Glu296 | [22] |
3 | MurC |  > >
| Tyr154, Asp174, His175, Arg298, Tyr313, His315, Thr344, His343,
Arg347, Ala417 | [23] |
4 | MurD | 
| Leu17, Lys19, Ser20, Gly21, Asn38, Thr330, Arg355, Ser424,
Phe431 | [24] |
5 | MurE* | 
| Ser30, Tyr45, Val47, Asn151, Thr152, Thr153, Ser179, Arg187, Arg383, Asp406,
Ser456, Glu460 | [25] |
6 | MurF | 
| Ile31, Phe45, Glu48, Asn49, Phe55, Ile145, Asn336, Ser338, Thr340, Glu368,
Asn369 | [26] |
7 | MraY | 
| Asp117, Asp118, Asp265, His324,
His325, His 326 | [27] |
8 | MurG | 
| Asn128, Arg164, Ile245, Ala264, Gln288, Gln289,
Glu269 | [8] |
9 | FemX/Fm
hB | 
| Lys36, Gly40, Tyr41, Leu103,
Tyr224, Tyr336 | [9] |
10 | FemA* | 
| Lys36, Gly40, Tyr41, Leu103, Tyr220, Phe224,
Ala326 | [9] |
11 | FemB | 
| Arg36, Gly40, Phe41, Leu103, Lys220, Tyr224,
Ser326 | [9] |
12 | PBP2*# | 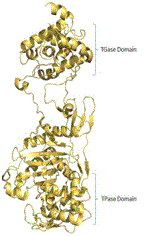
| Glu114, Glu144,
Ala146, Gln152, Lys155, Asp156, Lys163, Arg167, Lys168, Glu171, Tyr196, Pro231, Gln 232, Val233,
Asn235 (TGase
Domain) | [28] |
Ser398, Ser399, Leu400, Lys401, Ser454, Phe455, Asp456, Lys583,
The584, Gly585 (TPase domain residues) |
Yellow and green ribbon indicates the template and target protein structure respectively. *Proteins, for which crystal structure is available. #Protein contains two catalytic domains (TGase and TPase domains).
MD Simulations for the Docked Complexes
The best G-scored protein-ligand complexes were further evaluated for their structural stability in dynamic condition through MD simulations approach in GROMACS 4.5.5 [20]. Protein-ligand complex was solvated (cubic box) in SPC216 water model and ions were added for neutralization. Finally minimized (Steepest descent = 500 steps followed by conjugate gradient = 500 steps) and equilibrated (NVT and NPT for 1000 ps) system was taken for MD simulation (15000 ps) at constant temperature (300 K) and pressure (1 atm).
Three-dimension structure of the modelled proteins
The 3-D structures of modelled proteins generated five models for each of the proteins, however best model was selected based on the lowest DOPE value (Table 2) and validated using SAVES (Table 2), which indicated the predicted model to well correlate with the structural features defined in its sequence and allowed for further study (Table 3) depending on their reliability.
Binding affinity analysis
The interaction affinity of Rutin with other peptidoglycan synthesizing enzymes (Table 4) revealed Mur ligases to obtain G-score ranging between -4.585 to -7.088, however, affinity of Rutin towards MurD was more (G-score, -7.088) in comparison to other Mur ligases, which formed six H-bonds with bond lengths ranging from 1.8296 to 2.32826 Å, while MurF exhibited lowest affinity (-4.585) by forming three H-bonds (bond lengths between 1.99531 to 2.30373 Å). Similarly, homologous peptidyl transferases (FemX, A, and B) interacted with Rutin producing G-scores of -6.857, - 5.973, and -5.526 each by making three, seven and six H-bonds respectively. Interestingly, transmembrane protein MraY interacted well (five H-bonds with the bond lengths of 1.82063 to 2.48561 Å) with -5.345 G-score.
The interaction of Rutin with both TGase and TPase domains of PBP2, although revealed highest G-scores (-8.297 and -8.057) compared to other proteins, the scores were approximately same with major differences mediated by the number of H-bonds (Figure 2), each with ten and two (bond lengths from 1.8878 to 2.32564 Å and 1.94745 and 1.94973 Å respectively). Thus, based on this observation, further these complexes were compared with the complexes formed with the reference molecules (inhibitor) of Moenomycin and Ceftobiprole (Table 5, Figure 3). Although, TGase domain interacted with Rutin and Moenomycin (-8.297 and -8.0137 G-scores), a greater number of H-bonds (ten) were formed with rutin compared to Moenomycin (eight). However, interacting residues (as Lys163, Gln232 and Asp156) were common in both suggesting the binding efficiency of Rutin similar to the reference compound. In addition, π-π, π-cation and π-sigma interactions observed showed Rutin to form four π-cation interactions (one with Lys155 and Lys168 and two by Lys163), which was significantly absent in Moenomycin. On the other hand unlike TGase, TPase domain exhibited interaction with the reference compound Ceftobiprole and comparison with Rutin complexes, although revealed G- scores to be on par with each other (-7.924 and -8.057), Rutin complex exhibited only two H-bonds proving it less efficient as the key residue Ser398 that interacted with Ceftobiprole (2.0567 Å) and their π-π interactions were unobserved. Thus, the studies indicated the affinity of Rutin towards TGase than TPase domain. Interestingly, Rutin followed the same kind of interaction pattern in the TGase domain and interacted more efficiently compared to Moenomycin while, its interaction was very poor with TPase domain compared to Ceftobiprole. Hence, in order to ascertain the reasons for these variations, all these complexes were further analysed under dynamic conditions.
Here (A) represent PBP2 structure with the two domains TGase and TPase shown as cartoon; (B) Moenomycin, (C) Rutin, (D) Ceftobiprole and (E) Rutin interaction provided the details of the binding mode with TGase and TPase domain of PBP2 respectively. White ribbon represents the protein backbone. Pink, cyan and green stick represents Moenomycin, Ceftobiprole and Rutin respectively where, blue dotted line indicates H-bonds.
Evaluation of protein-ligand complex stability through MD simulation
The stability and behavior of Rutin, Moenomycin and Ceftobiprole complexed with both domains of bound PBP2 and unligated PBP2 were assessed by simulating for 15000 ps and analyzed by plotting the backbone root mean deviation (RMSD), root mean fluctuation of protein backbone (RMSF), radius of gyration (Rg) and H-bonds graph for the period of 15000 ps.
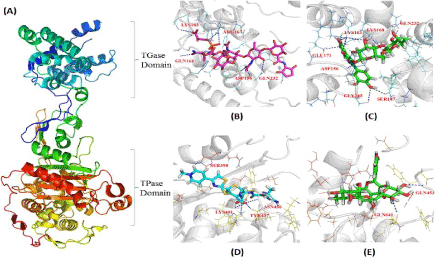
Figure 2.Comparative H-bond interaction pattern in the TGase and TPase domains of PBP2
Here (A) represent PBP2 structure with the two domains TGase and TPase shown as cartoon; (B) Moenomycin, (C) Rutin, (D) Ceftobiprole and (E) Rutin interaction provided the details of the binding mode with TGase and TPase domain of PBP2 respectively. White ribbon represents the protein backbone. Pink, cyan and green stick represents Moenomycin, Ceftobiprole and Rutin respectively where, blue dotted line indicates H-bonds.
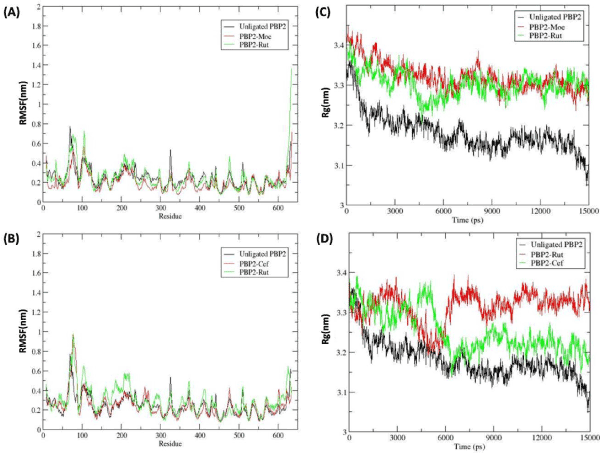
Figure 3. The RMSF and Radius of gyration plots represents the stability of the Protein-ligand Complexes
Table 4. Interaction pattern of the Rutin with the proteins involved in peptidoglycan synthesis
Proteins | G-score, Kcal/mol (Rutin) | Residues involved in H-bond | No of H-bond |
MurA | -6.461 | ARG95, ALA96, ASP308, ASP308, ASN23, ASN23 | 6 |
MurB | -5.394 | GLU72, GLU72, GLU171, VAL65 | 4 |
MurC | -5.417 | SER12, TYR313, ALA417, GLU150, GLU150 | 5 |
MurD | -7.088 | ASN145, SER168, ASN78, GLU166, LYS328, GLY80 | 6 |
MurE | -5.913 | ARG187, ASN407, HIS181, TYR351, GLU382, ALA150 | 6 |
MurF | -4.585 | SER33, SER33, SER33 | 3 |
MraY | -5.345 | SER108, ASN166, SER108, GLN269, GLN269 | 5 |
MurG | -5.583 | ARG260, SER263, ARG287, GLY282, ASP125 | 5 |
FemA | -5.973 | LYS33, LYS33, GLN154, ARG228, GLU36, VAL152, TYR69 | 7 |
FemB | -5.526 | LYS106, GLN155, THR152, TYR70, SER153, ASP37 | 6 |
FmhB/X | -6.857 | LYS374, LEU319, GLY356 | 3 |
Table 5. Differences in the docking pattern of Rutin at TPase and TGase domain of PBP2
PBP2-Lig Complex | G-score (Kcal/mol) | H-bond Residues | Bond Length (Ǻ) | Donor Atom | Acceptor Atom | No of H-bond |
TGase-Rutin | 8.297 | GLY145
SER147
LYS163
LYS163
LYS163
LYS168
GLN232
GLU171
ASP156
ASP156 | 1.8878
2.32564
2.24244
2.07449
2.04573
2.21883
2.05424
1.88623
1.80934
1.45512 | H
HG
HZ1
HZ2
HZ2
HZ3
H22
H23
H29
H30 | O4
O8
O12
O5
O10
O9
OE1
OE1
OD2
OD1 | 10 |
Tgase-Moenomyc | 8.0137 | GLN161
LYS163
ARG167
ARG167
ARG167
GLN232
ASP156
ASP156 | 1.74209
2.30966
2.38802
2.19337
2.16287
2.37979
2.34513
2.47206 | HE22
HZ3
HH12
HH22
HH22
H123
H97
H98 | O77
O14
O29
O29
O39
O
OD1
OD2 | 8 |
TPase-Rutin | 8.057 | GLN641
GLN453 | 1.94745
1.94973 | H23
H24 | O
OG1 | 2 |
|
TPase-Ceftobiprole | -7.924 |
LYS401
LYS401
SER398*
TYR437
ASN456 |
2.39515
2.4851
2.0567
1.97877
2.09275 |
HZ1
HZ2
HG
H
HD22 |
O3
O3
O9
O5
O1 | 5 |
*Key residue, Common interacting residues in comparison to the reference molecule (Moenomycin and Ceftobiprole) are bold-faced.
RMSD
The RMS deviation plots of PBP2 backbone (Figure 3A,3B) revealed TGase domain of bound form with Rutin and Moenomycin to deviate more compared to the unligated PBP2. The RMS deviation plot shows that the unligated PBP2 equilibrated after ~6000 ps and thereafter deviated between ~0.21 to ~0.31 nm. Whereas, backbone deviation was observed more in reference complex PBP2-Moenomycin and PBP2-Rutin in comparison to the unligated PBP2. The PBP2-Moenmycin complex reached at equilibrium after ~9000 ps and deviated between ~0.3 to ~0.4 nm while, PBP2-Rutin complex equilibrated from ~4000 ps and deviated between ~0.6 to ~0.7 nm. Noticeably, the deviation pattern in PBP2-Moenomycin and PBP2-Rutin was observed to be similar. On the other hand, when TPase domain was occupied by Rutin and Ceftobiprole the RMS deviation plot (Fig. 3B) showed less deviation in comparison to the TGase bound PBP2 complexes (PBP2-Rutin and PBP2-Moenomycin). The PBP2-Ceftobiprole equilibrated after ~2000 ps and further deviated between ~0.2 to ~0.3 nm, which was similar as the unligated PBP2. Whereas, PBP2-Rutin complex reached at equilibrium after ~8000 ps and thereafter deviated between ~0.3 to ~0.4 nm, which was more in comparison to the reference complex (Ceftobiprole) and unligated PBP2.
RMSF
In order to analyze the residual atomic fluctuations of the backbone atoms in the presence of Rutin at TGase and TPase domain, RMSF plots were generated for all complexes. The RMS fluctuation of individual amino acid in both domain complexes, which is shown in figure 3A,3B, revealed that the major movement took place at TGase domain which is present at the N-terminal region, with respect to the TPase domain. Comparative analysis of RMS fluctuation for both domain complexes (TGase and TPase complexes) showed that in the presence of ligand (Moenomycin and Rutin) at Tgase domain (between 100 to 250 residues) (Figure 3A) backbone fluctuation was less in comparison to the Tpase occupied complexes (Figure 3B). On the other hand, TPase domain, which is present at C-terminal (350 to 600 residues), was observed to be stable in both complexes. Based on the fluctuation pattern of PBP2 backbone in both complexes it was suggested that the Rutin stability at TGase domain was more in comparison to the TPase domain.
Radius of gyration
Further, to analyze the compactness TGase and TPase domain of PBP2 backbone in the presence of Rutin with respect to the Moenomycin and Ceftobiprole, Rg graph was plotted (Figure 3) and analyzed. The Rg values over the simulation time scale for both domains in ligand bound state were analyzed simultaneously. From the figure 3A, it was observed that the PBP2 backbone in the presence of the ligand (Rutin and Moenomycin) in the TGase domain showed more fluctuation in comparison to the unligated PBP2, where TGase-Moenomycin complex fluctuated least from ~3.4 to ~3.3 nm. The TGase- Rutin complex fluctuation was observed to decrease upto ~3.2 nm at ~4500 ps but thereafter increased and maintained in between ~3.4 to ~3.3 nm, which was similar as the reference complex (PBP2-Moenomycin). While, the comparative analysis of TPase domain Rg value showed more fluctuation in TPase-Rutin and TPase-Ceftobiporle Complex with respect to the unligated PBP2, where TPase-Ceftobiprole complex stabilize after ~7500 ps between ~3.4 to ~3.3 nm and TPase-Rutin complex was observed to be unstable for over the period of 15000 ps. Overall, Rg plot analysis showed that TPase domain fluctuation was more in comparison to the TGase domain. Based on the Rg plot, it was clear that TGase-Rutin Rg curves significantly followed the reference complex fluctuation pattern with respect to the TPase domain complexes, suggesting Rutin was able to stabilize the PBP2 backbone in TGase bound state (Figure 3C,3D).
Hydrogen-bonds
The stabilization of protein–ligand complexes also depends on the intermolecular H-bonds, therefore the stability of the H-bonds formed at TGase and TPase domain of PBP2 with Rutin and their inhibitors (Moenomycin and Ceftobiprole respectively) throughout the simulation were computed and shown in figure 4. The H-bond plot showed Rutin formed three and one H-bonds (shown as red in the Figure 4A,4B) at TGase and TPase domains respectively while the reference compound Moenomycin and Ceftobiprole formed ~4 and ~3 H-bonds at TGase and TPase domain respectively throughout the simulation period (Figure 4C,4D). Here, (A) and (B) represents the RMSD of PBP2 (C) represent Moenomycin and Rutin while (D) Ceftobiprole and Rutin interaction pattern in TGase and TPase domain respectively during the simulation period (15000 ps).
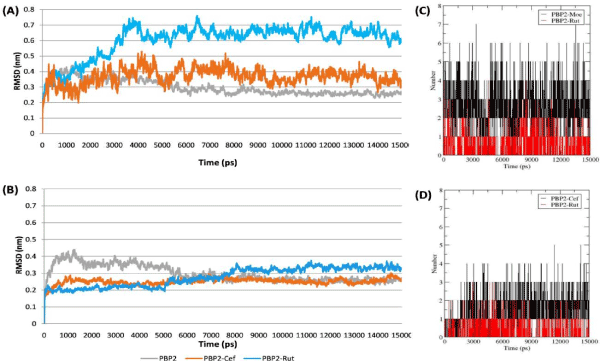
Figure 4. The plots represent the stability of the Protein-ligand Complexes mediated by the Hydrogen bond4
The previous study on the Rutin binding affinity towards the PBP2a active site [29,30], provided the information of Rutin efficiency against MRSA. In order to investigate its capability towards the other peptidoglycan synthesizing enzymes, molecular docking and simulation studies were performed and analyzed. Enzymes MurA, MurB, MurC, MurD, MurE, MurF, MurG, MraY, Fem X, Fem A, Fem B, and PBP2 were found to be involved in peptidoglycan synthesis [31]. Among them, only MurB, MurE, FemA, and PBP2 proteins crystal structure were available in the PDB, hence other proteins tertiary structures were modeled and the lowest potential energy (PE) conformations were selected for further studies. Using molecular docking approach, it was observed that the Mur ligases, trans membrane protein MraY, and homologous peptidyl transferases (FemX, FemA, and FemB) active site interacted with Rutin by obtaining G-score in between -4.585 and -7.088, which was observed to be less in comparison to the PBP2.
Studies has revealed that S. aureus contains four types of PBPs namely PBP1 [32], PBP2 [33], PBP3 [34]and PBP4 [35] and among them PBP2 was observed to be essential for the cell viability in sensitive S. aureus strain [33]. Since PBP2 catalyzes two important steps (transglycosylation and transpeptidation) of peptidoglycan synthesis with the help of two different catalytic sites namely TGase and TPase domain, hence Rutin affinity was analyzed for both of the domains. The molecular docking studies showed that the Rutin G-score for both domains (TGase and TPase) was approximately similar, but interestingly Rutin formed ten (bond length ranged between 1.8878 to 2.32564 Å) and two H-bonds (1.94745 and 1.94973 Å) at TGase and TPase domain respectively. Therefore, interaction pattern of TGase and TPase docked Rutin complexes were analyzed by comparing with the reference molecule Moenomycin (Moe) and Ceftobiprole (Cef) respectively, which were known for their activity against S. aureus. Moenomycin is a natural antibiotic that inhibits transglycosylation step of bacterial cell wall synthesis by binding to the TGase domin of PBP2 [36], whereas Ceftobiprole is known for inhibiting the transpeptidation step in cell wall synthesis [37]. However, their docking results (Table 4 and shown in Figure 3,4) showed that Rutin and Moenomycin interacted with -8.297 and -8.0137 of G-score respectively. Moreover, Lys163, Gln232 and Asp156 were observed to be common in both interactions, also these residues were observed in PBP2 crystal structure [28]. However, in case of TPase domain, Rutin and Ceftobiprole G-score were -8.057 and -7.924 but Rutin formed only two H-bonds while Ceftobiprole interacted by making five bonds. The crystallographic study [28] of PBP2 revealed the importance of Ser398 in TPase domain, which is required for its catalytic
activity was observed to be absent in Rutin interaction. Based on the docking result, it was concluded that the Rutin binding affinity towards the TGase domain was more in comparison to the TPase domain.
In order to investigate more about these variations and to observe their interaction in the dynamic condition both complexes were further simulated for 15000 ps in dynamic condition. Simulation results were analyzed by plotting the RMS deviation, RMS fluctuation, and radius of gyration plot through comparing with reference complex (Moenomycin and Ceftobiprole bound TGase and TPase respectively) and unligated PBP2. However, RMS deviation and Rg plot showed PBP2 backbone was stabilized between 0.6 to 0.7 nm and (Figure 4A) and ~3.4 to ~3.3 nm (Figure 3A) respectively, which was more in comparison to the other complexes but, the deviation pattern was observed to be similar as reference complex (TGase-Moenomycin). While in case of TPase-Rutin bound complexes, backbone deviation (Figure 4B) and fluctuation (Figure 3b) pattern was found to be different than references complexes. These observations affirm the Rutin capability towards TGase domain in comparison to the TPase domain.
Finally, from the present study it was concluded that the Rutin was unable to interact efficiently with other cell wall synthesizing enzymes (MurA, MurB, MurC, MurD, MurE, MurF, MurG, MraY, FemX, FemA, FemB), but interacted well with PBP2. Interestingly, Rutin interaction potential towards the TGase domain was observed to be better and similar as Moenomycin than TPase domain. However, Rutin interacted to the TPase domain also but number of H-bonds and other interactions (π-interactions, Hydrophobic, Van Der Waals) including the Ser398 interaction were observed to be missing. In addition, MD simulation validated the Rutin stability in TGase domain based on the PBP2 backbone deviation and fluctuation. Overall, these results strongly suggested that Rutin can be further modified and explored as a potential next generation antibiotic development against S. aureus.
- Silhavy TJ, Kahne D, Walker S (2010) The bacterial cell envelope. Cold Spring Harb Perspect Biol 2: a000414.[Crossref]
- Ghuysen JM, Hakenbeck R (1994) Bacterial cell wall. (1st Edn), Elsevier.
- van Heijenoort J (1998) Assembly of the monomer unit of bacterial peptidoglycan. Cell Mol Life Sci 54: 300-304. [Crossref]
- Blake KL, O'Neill AJ, Mengin-Lecreulx D, Henderson PJ, Bostock JM, et al. (2009) The nature of Staphylococcus aureus MurA and MurZ and approaches for detection of peptidoglycan biosynthesis inhibitors. Mol Microbiol 72: 335-343. [Crossref]
- Patin D, Boniface A, Kovač A, Hervé M, Dementin S, et al. (2010) Purification and biochemical characterization of Mur ligases from Staphylococcus aureus. Biochimie 92: 1793-1800. [Crossref]
- Lovering AL, Safadi SS, Strynadka NC (2012) Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem 81: 451-478. [Crossref]
- Bouhss A, Crouvoisier M, Blanot D, Mengin-Lecreulx D (2004) Purification and characterization of the bacterial MraY translocase catalyzing the first membrane step of peptidoglycan biosynthesis. J Biol Chem 279: 29974-29980. [Crossref]
- Hu Y, Chen L, Ha S, Gross B, Falcone B, et al. (2003) Crystal structure of the MurG: UDP-GlcNAc complex reveals common structural principles of a superfamily of glycosyltransferases Proc Natl Acad Sci 100: 845-849. [Crossref]
- Biarrotte-Sorin S, Maillard AP, Delettré J, Sougakoff W, Arthur M, et al. (2004) Crystal structures of Weissella viridescens FemX and its complex with UDP-MurNAc-pentapeptide: insights into FemABX family substrates recognition. Structure 12: 257-267. [Crossref]
- Benson TE, Prince DB, Mutchler VT, Curry KA, Ho AM, et al. (2002) X-ray crystal structure of Staphylococcus aureus FemA. Structure 10: 1107-1115. [Crossref]
- Schneider T, Senn MM, Berger-Bächi B, Tossi A, Sahl HG, et al. (2004) In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II‐Gly5) of Staphylococcus aureus. Mol Microbiol 53: 675-685. [Crossref]
- Navarre WW, Schneewind O (1999) Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol Mol Biol Rev 63: 174-229. [Crossref]
- Hao H, Cheng G, Dai M, Wu Q, Yuan Z (2012) Inhibitors targeting on cell wall biosynthesis pathway of MRSA. Mol Biosyst 8: 2828-2838. [Crossref]
- Webb B, Sali A (2014) Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics 47: 1-32. [Crossref]
- Eramian D, Shen MY, Devos D, Melo F, Sali A, et al. (2006) A composite score for predicting errors in protein structure models. Protein Sci 15: 1653-1666. [Crossref]
- MacArthur MW, Laskowski RA, Thornton JM (1994) Knowledge-based validation of protein structure coordinates derived by X-ray crystallography and NMR spectroscopy. Curr Opin Struct Biol 4: 731-737.
- Lüthy R, Bowie JU, Eisenberg D (1992) Assessment of protein models with three-dimensional profiles. Nature 356: 83-85. [Crossref]
- Nagpal I, Raj I, Subbarao N, Gourinath S (2012) Virtual screening, identification and in vitro testing of novel inhibitors of O-acetyl-L-serine sulfhydrylase of Entamoeba histolytica. PLoS One 7: e30305. [Crossref]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, et al. (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49: 6177-6196. [Crossref]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, et al. (2005) GROMACS: fast, flexible, and free. J Comput Chem 26: 1701-1718. [Crossref]
- Yoon HJ, Lee SJ, Mikami B, Park HJ, Yoo J, et al. (2008) Crystal structure of UDP‐N‐acetylglucosamine enolpyruvyl transferase from Haemophilus influenzae in complex with UDP‐N‐acetylglucosamine and Fosfomycin. Proteins 71: 1032-1037. [Crossref]
- Benson TE, Harris MS, Choi GH, Cialdella JI, Herberg JT, et al. (2001) A structural variation for MurB: X-ray crystal structure of Staphylococcus aureus UDP-N- acetylenolpyruvylglucosamine reductase (MurB). Biochemistry 40: 2340-2350. [Crossref]
- Kurokawa K, Nishida S, Ishibashi M, Mizumura H, Ueno K, et al. (2008) Staphylococcus aureus MurC participates in L-alanine recognition via histidine 343, a conserved motif in the shallow hydrophobic pocket. J Biochem 143: 417- 424. [Crossref]
- Tomasić T, Zidar N, Sink R, Kovac A, Blanot D, et al. (2011) Structure-based design of a new series of D-glutamic acid-based inhibitors of bacterial UDP-N-acetylmuramoyl-L-alanine: D-glutamate ligase (MurD). J Med Chem 54: 4600-4610. [Crossref]
- Ruane KM, Lloyd AJ, Fülöp V, Dowson CG, Barreteau H, et al. (2013) Specificity determinants for lysine incorporation in Staphylococcus aureus peptidoglycan as revealed by the structure of a MurE enzyme ternary complex. J Biol Chem 288: 33439-33448. [Crossref]
- Hrast M, Turk S, Sosič I, Knez D, Randall CP, et al. (2013) Structure–activity relationships of new cyanothiophene inhibitors of the essential peptidoglycan biosynthesis enzyme MurF. Eur J Med Chem 66: 32-45. [Crossref]
- Chung BC, Zhao J, Gillespie RA, Kwon DY, Guan Z, et al. (2013) Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science 341: 1012-1016. [Crossref]
- Lovering AL, de Castro LH, Lim D, Strynadka NC (2007) Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis, Science, 315: 1402-1405. [Crossref]
- Rani N, Vijayakumar S, P T V L, Arunachalam A (2015) Allosteric site mediated active site inhibition of PBP2a using Quercetin 3-O-Rutinoside and its combination. J Biomol Struct Dyn 34: 1-35. [Crossref]
- Rani N, Vijayakumar S, Thanga Velan LP, Arunachalam A (2014) Quercetin 3-O-rutinoside mediated inhibition of PBP2a: computational and experimental evidence to its anti-MRSA activity. Molecular BioSystems 10: 3229-3237. [Crossref]
- Bugg TD, Braddick D, Dowson CG, Roper DI (2011) Bacterial cell wall assembly: still an attractive antibacterial target. Trends Biotechnol 29: 167-173. [Crossref]
- Pereira SF, Henriques AO, Pinho MG, de Lencastre H, Tomasz A (2009) Evidence for a dual role of PBP1 in the cell division and cell separation of Staphylococcus aureus. Mol Microbiol 72: 895-904. [Crossref]
- Pinho MG, de Lencastre H, Tomasz A (2001) An acquired and a native penicillin-binding protein cooperate in building the cell wall of drug-resistant staphylococci. Proc Natl Acad Sci 98: 10886-10891. [Crossref]
- Pinho MG, de Lencastre H, Tomasz A (2000) Characterization, and Inactivation of the Gene pbpC, Encoding Penicillin-Binding Protein 3 of Staphylococcus aureus. J Bacteriol 182: 1074-1079.
- Navratna V, Nadig S, Sood V, Prasad K, Arakere G, et al. (2010) Molecular basis for the role of Staphylococcus aureus penicillin binding protein 4 in antimicrobial resistance. J Bacteriol 192: 134-144. [Crossref]
- Taylor JG, Li X, Oberthür M, Zhu W, Kahne DE (2006) The total synthesis of moenomycin A. J Am Chem Soc 128: 15084-15085. [Crossref]
- Bush K, Heep M, Macielag MJ, Noel GJ (2007) Anti-MRSA β-lactams in development, with a focus on ceftobiprole: the first anti-MRSA β-lactam to demonstrate clinical efficacy. Expert Opin Investig Drugs 16: 419-429. [Crossref]