Pancreatic ductal adenocarcinoma (PDAC) arises from epithelia of pancreas. Despite its low incidence, it is the most lethal cancer type. Although the poor outcome is largely secondary to the high proportion of patients who are diagnosed with advanced disease, the prognosis of PDAC is also influenced by the inherent biological aggressiveness and the high metastatic potential of this malignancy. Treatment options remain limited with little progress over the last decades. Some improvements in surgical outcome occur in patients who also receive chemotherapy and/or radiotherapy, however, the impact on long-term survival has been minimal owing to the intense resistance of PDAC to all extent treatments regimen. Hence, there is an urgent need to 1) gain better understanding of the biology of PDAC; 2) to develop early detection and prevention programs; 3) to identify new therapeutic strategies to improve quality of life and survivorship. In this review, first, we will summarise the state of knowledge of PDAC pathogenesis with a particular the focus on the molecular characteristics causing therapeutic resistance. Then, we will briefly review current and emerging approaches in the PDAC care. Lastly, we will highlight the integrative approaches in the light of new experimental and clinical research conducted with the aim of moving towards personalised therapy in patients with PDAC.
pancreatic ductal adenocarcinoma, personalised medicine, integrative oncology
PDAC is among the most lethal cancer with an incidence rate equalling that of its mortality rate [1,2]. PDAC is the twelfth most common cancer in the world, and is the seventh most common cause of death from cancer [3,4]. The highest incidence and mortality rates of PDAC are found in the Western world [4]. For both sexes, there has been a steady increase in incidence over the past 20 years [3,5]. The dismal outlook of this disease is mostly due to the majority (~80 %) of patients being diagnosed with advanced-stage disease, with severe cachexia and poor metabolic status rapidly contributing to morbidity and mortality [6]. Currently, there is no effective screening and no early detection method available to diagnose PDAC at a pre-malignant stage. Although surgery to remove pancreatic tumours offers the best chance for survival, only a minority (10-15 %) of patients can undergo curative operation at the time of diagnosis [6]. Even after surgery, the overall 5-year survival rate is less than 4% mainly because micrometastases will already have been established, eventually leading to local and/or systemic recurrence [3]. Some improvements in surgical outcome can occur in patients who also receive chemotherapy and/or radiotherapy, however, the impact on long-term survival has been minimal owing the intense resistance of the disease to current treatment regimens. Consequently, new strategies are urgently needed for its clinical management.
In this review, first, we will summarise the state of knowledge of PDAC pathogenesis with a particular the focus on the molecular characteristics causing therapeutic resistance. Then, we will briefly review current and emerging approaches in the PDAC care. Lastly, we will highlight the integrative approaches in the light of new experimental and clinical research conducted with the aim of moving towards personalised therapy in patients with PDAC.
Pancreas is an elongated and lobular abdominal organ, containing blood and lymphatic vessels, nerves and excretory ducts. It can be divided into four sections: the head, neck, body and tail (Figure 1a). The wider end of the pancreas, close to the duodenum, is referred to as the head, the middle portion is called the body and the rest, the tail, extends to the hilum of the spleen. Being fixed within the retroperitoneum and having intimate relations with the duodenum, common bile duct, stomach, transverse colon, left kidney, left adrenal gland and spleen means that invasiveness can occur readily in many directions (Figure 1a).
Pancreas plays a dual role as an organ of both the digestive and the endocrine systems. The endocrine pancreas regulates metabolism and glucose homeostasis through the secretion of hormones. It consists of clusters of cells known as islets of Langerhans that are categorized by their secretory function: Beta cells produce insulin, alpha cells produce glucagon, delta cells produce somatostatin and PP cells produce pancreatic polypeptide hormone (Figure 1b). The exocrine pancreas consists of acinar, ductal and centroacinar cells (Figure 1c). While acinar cells synthesize digestive enzymes packed into zymogen granules, ductal cells produce alkaline fluid that is rich in sodium bicarbonate ions (NaHCO3) and mucus to flush the zymogens into the intestine [7]. Under physiological conditions, NaHCO3 secretion neutralises the acid secreted by acinar cells to prevent aggregation of digestive enzymes in the lumen and to neutralise the acid chyme entering the duodenum from the stomach [7]. However, under pathophysiological conditions, enhanced and prolonged acidification can cause obstruction of the duct lumen by precipitating proteins and/or viscous juice and disrupting intercellular junctions, ultimately contributing to possible pancreatitis, which is a well-known risk factor for PDAC [7,8]. The endocrine and exocrine functions of the pancreas are regulated by an integrated system of neural input and hormonal mechanisms.
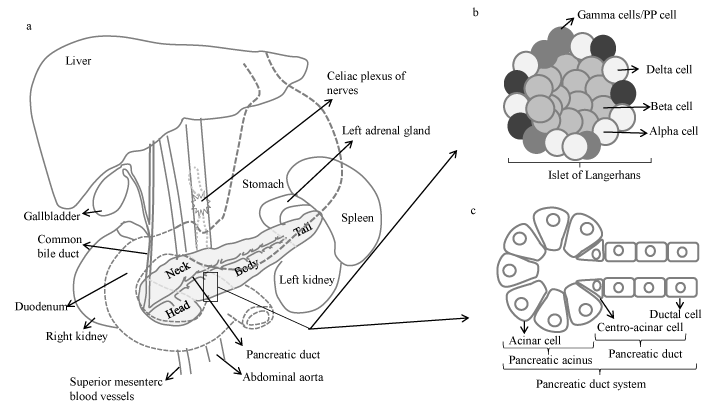
Figure 1. a)Gross anatomy of pancreas and its location in the abdominal cavity. Illustration of b) islet of Langerhans, and c) pancreatic duct system.
Although the exact causes of PDAC are not known, epidemiological studies have established both non-modifiable (inherited) and modifiable (non-inherited) factors as contributing to disease development (Figure 2). The most recent Surveillance, Epidemiology and End Results Program (SEER) and the National Center for Health Statistics data indicate that ~1.5 % of men and women will be diagnosed with PDAC during lifetime [9]. However, currently, there is no accepted standard for evaluating, screening or stratifying such high-risk patients or preventing disease occurrence. The reasons for the increase in incidence are not clear but, at the increased incidence of obesity is likely a contributory factor [10]. In addition, metabolic syndrome, diabetes, older age, smoking, alcohol abuse, high-fat diets, certain trace elements, male gender, non ‘0’ blood type and African-American ethnicity are among the additional risk factors (Figure 2) [10,11]. Chronic pancreatitis is associated with an increased risk of PDAC; the link between chronic pancreatitis and PDAC is strongest in smokers or a group with hereditary chronic panreatitis [12] Several studies reported increased PDAC risk among people with chronic hepatitis B, hepatitis C, HIV, and Helicobacter pylori (H. pylori) infections [10,11]. In addition, a history of cholecystectomy or partial gastrectomy and periodontal disease were associated with increased PDAC risk [10,11]. Approximately, 5-10% of PDACs have familial basis, falling into a category of Familial Pancreatic Cancers (FPC) [12]. However, only a minority (around 20%) of FPC has been linked to known genetic syndrome or causal gene mutation (Table 1) [12]. This implies that PDAC does not generally follow Mendelian inheritance and its development is largely contignent on the independent and interactive effects of genes and environment. In this regard, it has been reported that smoking triples the risk of PDAC in members of FPC families [12].
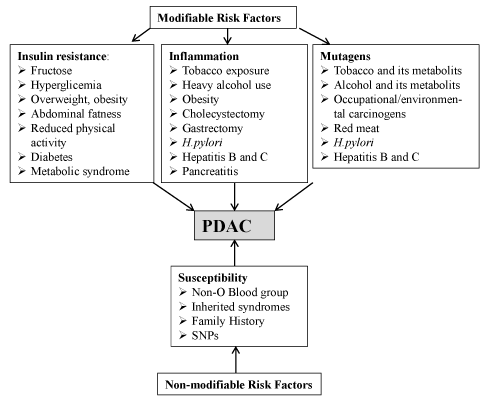
Figure 2. Risk factors. (Abbreviations: SNP, single nucleotide polymorphism; Helicobacter pylori: H. pylori)
Cancers of the pancreas fall into two groups: exocrine and endocrine. More than 95% of all pancreatic cancers originate in the exocrine pancreas and of these, ~ 90 % constitute PDACs [2]. About 65% of PDACs develop in the head of the pancreas, 30 % in the body and tail, and 5% can involve the whole pancreas [2].
Development of PDAC is described classically by morphological and molecular transformation from precursor lesions leading to invasive carcinoma (Figure 3). Genetic studies have established that PDAC can have presursor lesions, termed PanIN (pancreatic intraepithelial neoplasia), IPMN (intraductal papillary mucinous neoplasm) and MCN (mucionous cystic neoplasm) arising from ductal epithelia of pancreas [2,13]. However, recent evidence emerging from mouse models and lineage tracing studies have also suggested that PDAC can also develop in the centroacinar-acinar compartment through a process of acinar-ductal metaplasia or through the expansion of the centroacinar cells accompanied by apoptosis of the acinar cells [13]. The majority of PDACs develop from PanINs (sub- PanIN1-3), representing increasing hyperplasia and cytological atypia characterised by loss of polarity, nuclear crowding, enlarged nuclei, pseudo-stratification and hyperchromatism [2,14]. PDAC grows in a disorganised pattern, infiltrates the pancreatic parenchyma, thus, the margins of the tumour are poorly defined [2,14]. One of the histological hallmarks of PDAC is a dense fibrotic stromal matrix, called desmoplasia, composed of extracellular matrix (ECM), mesenchymal cells, nerve cells, inflammatory cells, as well as blood and lymphatic vessels that together can comprise the bulk (up to 90%) of the tumour volume [15,16]. Interestingly, while PDAC elicits an intense desmoplastic reaction within the pancreas itself, desmoplasia may be weak or absent in metastatic foci [14]. Low vascular density is another characteristic of PDAC, hindering delivery of sufficient oxygen and nutrients and causing central necrosis in larger neoplasms [14]. Additionally, vascular and perineural invasion, and neural remodelling with enhanced neural density and hypertrophy are highly characteristic of PDAC [17]. Lymphatic invasion is another very common finding and is associated with lymph-node metastasis [2]. Cancer cells may invade the wall of blood vessels or penetrate the lumen causing thrombosis [2]. At the time of diagnosis, invasion into adjacent peripancreatic adipose tissue, bile duct, hepatopancreatic duct, and duodenal mucosa is common, causing obstruction of associated duct system [2].
PDAC arises from multiple spontaneous and/or inherited mutations and epigenetic alterations, which reflects on intracellular signalling pathways that normally control vital cellular events and the cellular response to extrinsic factors (Figure 3, 4). Given the dismal prognosis associated with PDAC, detailed understanding of the molecular mechanisms that stimulate the promotion and progression of sub-malignant cells into PDAC cells will most likely help to identify novel targets and agents for treatment and chemoprevention. Presented in this section is a review of the predominant molecules and signalling pathways that are deregulated in PDAC and associated with the tumour and/or the stromal compartment (Figure 4).
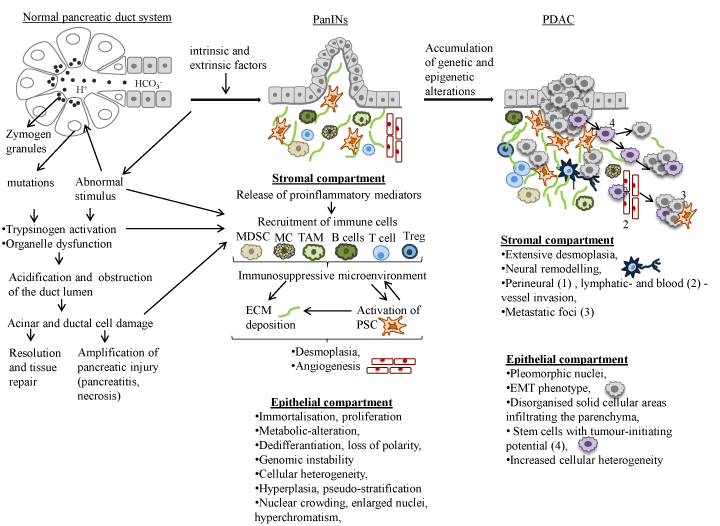
Figure 3. Progression model of PDAC from normal epithelium to invasively growing metastatic tumour. The progression is associated with the stepwise accumulation of specific genetic and epigenetic alterations in high-frequency driver genes. These histopathological changes are accompanied by infiltrating immune cells and an increasing desmoplastic stromal response. (Abbreviations: PRSS1,protease, serine, 1 (trypsin 1); SPINK1, serine peptidase inhibitor, kazal type 1; MC, mast cell; MDSC, myeloid-derived suppressor cell; TAM, tumour associated macrophage; Treg, regulatory T cell; PSC, pancreatic stellate cells; ROS, reactive oxygen species; MMP, matrix metalloproteinases; TIMP, tissue inhibitor of metalloproteinases; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition.
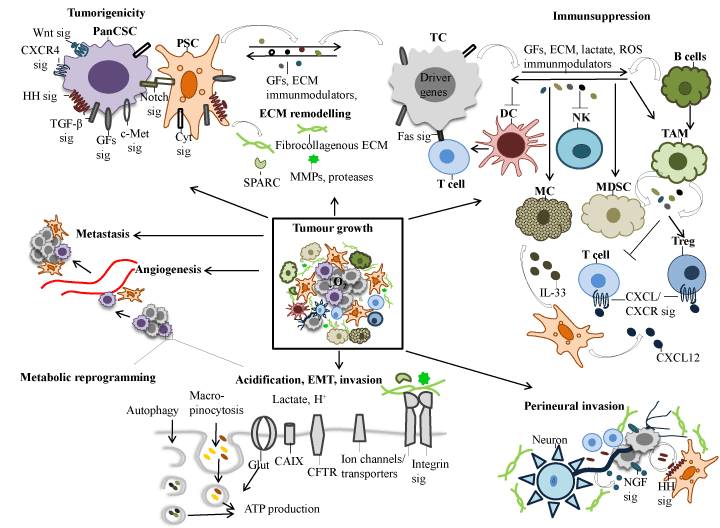
Figure 4. Schematic model summarising the main characteristics of PDAC. The critical molecular pathways leading to the development and progression of PDAC depicted in this figure is discussed in the text. Abbreviations: Sig, signalling; PancSC, pancreatic cancer stem cell; PSC, pancreatic stellatte cell; TGF-β, transforming growth factor-beta; GFs, growth factors, IGF, insulin-like growth factor, INS, insulin; EGF, epithelial growth factor; HGF, hepatocyte growth factor; VEGF, vascular epidermal growth factor; PDGF, platelet-derived growth factor; HH, hedgehog; Cyt, cytokines; MC, mast cell; MDSC, myeloid-derived suppressor cell; TAM, tumour associated macrophage; Treg, regulatory T cell; DC, dendritic cells ROS, reactive oxygen species; MMP, matrix metalloproteinases; SPARC, secreted protein acidic and rich in cysteine; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition; IL, interleukin; cyclooxygenase-2, COX-2; CXCL13, chemokine (C-X-C motif) ligand 13; CXCL12, chemokine (C-X-C motif) ligand 12; CXCR2, C-X_C motif chemokine receptor 2, CXCR4, C-X-C motif chemokine receptor 4; NGF, nerve growth factor; mtCFTR, mutant cystic fibrosis transmembrane conductance regulator; Glut, glucose transporter; CA IX, carbonic anhydrase; ATP, adenosine triphosphate; O2, oxygen; H+ , hydrogen ion; EMT, epithelial-mesenchymal transition.
PDAC is a polygenic disease with multiple high and low germline susceptibility alleles (Table 1). Genetic abnormalities in PDAC are complex, with multiple chromosomal losses and gains, various copy number alterations, microsatellite instability, and intragenic point mutations [18,19]. The first large sequencing study identified the oncogene, KRAS, and the tumour suppressor genes, CDKN2A, TP53, and SMAD4 as the four main ‘driver’ genes in PDAC [18]. Both sporadic and familial sporadic and familial PDAC share the same driver mutations found in KRAS, CDKN2A, TP53 and SMAD4 genes [20]. SMARCA4, CDH1, EPHA3, FBXW7, EGFR, IDH1, and NF1 mutations have been identified as low-frequency drivers [18]. Recently, a whole-genome sequencing and copy number variation analysis of 100 PDACs, have revealed additional candidate drivers, ARID1A, ROBO2, KDM6A and PREX2 [19]. These mutations were associated with 12 core signalling pathways, including those for apoptosis, DNA damage, KRas signalling, TGFB signalling, and epigenetic modification found to be targeted in more than thirds of the cancers evaluated (Table 2) [18,19]. Notably, detection of a long list of infrequent variations in chromosomal structure, many of which contained oncogenes (ERBB2, MET, FGFR1, CDK6, PIK3R3 and PI3CA) that may be druggable targets, also implies significant inter- and intra-tumoral heterogeneity [19]. Importantly, Waddell et al. [19] demonstrated that genomic instability co-segregated with inactivation of DNA maintenance genes, BRCA1, BRCA2 and PALB2, and a mutational signature of DNA damage repair deficiency.
Table 1. Suggested PDAC genetic risk factors [11].
Risk factors |
Gene |
Increased risk |
Breast and ovarian cancer syndromes |
BRCA2, BRCA1, PALB2 |
2-5 |
Familial atypical multiple mole melanoma |
CDKN2A |
47 |
Peutz-Jeghers |
STK11/LKB1 |
132 |
Hereditary nonpolyposis colorectal cancer |
MMRs (MSH2, MLH1, PMS1, PMS2, MSH6) EPCAM |
8.6 |
Familial adenomatous polyposis |
APC |
4.5-6 |
Hereditary pancreatitis |
PRSS1, SPINK1 |
69 |
Cystic Fibrosis |
CFTR |
3.5 |
Li-Fraumeni |
TP53 |
7.3(%) |
Ataxia-telangiectasia |
ATM |
Increased |
Non-O blood group |
|
1.3 |
Familial Pancreatic Cancer |
Unknown |
9 (1FDR)
32 (3FDRs) |
(FDR, first-degree relative )
Table 2: Core signalling pathways and processes genetically altered in the majority of pancreatic cancers [18,19].
Signalling Pathways and processes |
Genetically altered genes |
KRas signalling |
KRAS, MAP2K4, RASGRP3, PREX2 |
Wnt/Notch signalling |
MYC,PPP2R3A, WNT9A, GATA6, TCF4, MAP2, TSC2 |
Small GTPase-dependent signalling (other than KRas) |
AGHGEF7, AGHGEF9, CDC42BPA, DEPDC2, PLCB3, PLCB4, RP1, PLXNB1, PRKCG |
TGF-β signalling |
TGFBR2, BMPR2, SMAD4, SMAD3 |
c-Jun N-terminal kinase signalling |
MAP4K3, TNF, ATF2, NFATC3 |
Integrin signalling |
ITGA4, ITGA9, ITGA11, LAMA1, LAMA4, LAMA5, FN1, ILK |
Hedgehog signalling |
TBX5, SOX3, LPR2, GLI1, GLI3, BOC, BMPR2, CREBBP |
Regulation of invasion |
ADAM11, ADAM12, ADAM19, ADAM5220, ADAMTS15, DPP6, MEP1A, PCSK6, APG4A, PRSS23, ROBO2 |
Homophilic cell adhesion |
CDH1, CDH10, CDH2, CDH7, FAT, PCDH15, PCDH17, PCDH18, PCDH9, PCDHB16, PCDHB2, PCDHGA1, PCDHGA11, PCDHGC4 |
Regulation of G1/S phase transition |
CDKN2A, FBXW7, CHD1, APC2 |
DNA damage control |
ERCC4, ERCC6, EP300, RANBP2, TP53 |
Apoptosis |
CASP10, VCP, CAD, HIP1 |
KRas signalling pathway
Mutations in the oncogene KRAS are the earliest detectable genetic alteration found in >99% of Pan1N-1 lesions and found in nearly 100 % of PDAC [21,22]. Mutations in KRAS locks KRas in a permanently active state leading to constitutive activation of downstream signalling pathways, including B-raf/MAPK/ERK (extracellular signal-regulated kinase), the phosphoinositide-3-kinase (PI3K)/3-phosphoinositide-dependent protein kinase 1 (PDK1)/AKT kinase and Ral guanine nucleotide exchange factor [22]. KRAS-driven mouse studies showed that oncogenic KRAS (KRASG12D or KRAS G12V) is essential for initiation, progression and maintenance of PDAC and of metastatic lesions [23,24]. Introduction of an inactivating mutation in tumour suppressor genes CDKN2A, TRP53, DPC4/SMAD4 or activation mutations in BRAF, greatly accelerates PanINs and PDAC development in KRAS-driven genetically engineered animals [25,27]. However, the presence of oncogenic KRAS in normal tissues and benign diseases suggests that KRas activation alone is unlikely to single-handedly promote carcinogenesis [28]. Mutual interactions between inflammatory stimuli and KRas signalling is sufficient to drive development of full-spectrum PanIN, desmoplasia and invasive PDAC [29]. Mechanistically, KRas signalling upregulates Hedgehog signalling, generation of inflammatory mediators (such as nuclear factor-kappa-B (NF–κB), cyclooxygenase 2 (COX-2), signal transducer and activator of transcription 3 (STAT3)), which are known to mediate paracrine interactions between epithelial cells and their surrounding microenvironment [29,30].
Strikingly, under hypoxic/nutrient deprived conditions, oncogenic KRAS confers selective advantage to the mutated cells by reprogramming tumour metabolism to maintain growth and survival [31]. By doing so, KRas signalling promotes glycolysis, glutamine-driven oxidative phosphorylation, autophagy and macropinocytosis. KRas-driven autophagy provides additional sources of nutrients within cells, as well as protects cells from ROS-mediated damage, providing stress tolerance [32].
IGF/Insulin axis
Recalling the link between energy-dense diets, diet-related metabolic disorders (e.g. obesity and type 2 diabetes) and increased risk for development of PDAC, it is most likely that peripheral insulin resistance, compensatory overproduction of insulin and increased bioavailability of insulin-like growth factor 1 (IGF-1) are important elements in PDAC. Upregulated IGF-1 signalling is involved in development and progression of PDAC, through induction of glucose uptake, differentiation, migration, cell proliferation and survival [33]. In an orthotopic model, autocrine IGF-1/IGF-1R signalling leads to activation of PI3K/AKT signalling occurring downstream of oncogenic KRas/B-raf/ERK, playing a role in pancreatic tumour initiation [34]. Reciprocally, AKT signalling promoted the invasiveness of PDAC cells through the upregulation of IGF-IR expression [35]. v-AKT thymoma viral oncogene homolog 2 (AKT2) gene amplifications, overexpression and activation are observed in 10% to 20% of PDACs [36]. AKT activation is negatively controlled by Phosphatase and tension homolog (PTEN) that is downregulated possibly due to promoter hypermethylation [37]. Respectively, pancreas specific deletion of one copy of PTEN was shown to rapidly accelerated KrasG12D-driven PDAC [38]. Inactivation of the key tumour suppressor gene, p53, as seen during the progression of the most PDAC cases, also leads to upregulation of the IGF-1/AKT/mTOR pathway [39]. In addition to growth-promoting signalling, mTOR also mediates negative feedback loops that restrain signalling through inhibiting both activation and expression of insulin receptor substrate (IRS-1). IRS-1 transmits signals from insulin and IGF-1 to the PI3K/AKT and ERK/MAPK pathways [40].
IGF-1R may interact with the insulin receptor (IR), G protein-coupled receptors (GPCRs), the epithelial growth factor receptor (EGFR), MET and in so doing promotes PDAC [41-44]. Notably, the Insulin/IGF-1 receptor (IR/IGF-1R) system plays a critical role in PDAC development [41]. Due to its high homology, IR forms hybrids with IGF-1R [41]. The insulin receptor isoforms A and IGF-1R hybrids bind both insulin and IGFs with similar affinity, in particular at high concentrations of intra-pancreatic insulin [41,45]. The insulin receptor isoforms A (IR-A) that are usually found in foetal tissues, gradually increase from the stage of hyperplastic lesions to PDAC [45]. Overexpression of IR-A accelerates the growth pathway by various mechanisms including; i) IGF-II binding to IR-A, ii) IGF-II binding to IR-A/IR-B hybrids, iii) IGF-II binding to IR-A/IGF-IR [45].
STK11/LKB1-AMPK pathway
Serine/Threonine Kinase 11, Liver Kinase B1 (STK11/LKB1) inactivating mutations appears both in familial and sporadic PDAC [12,47]. In a mouse model, LKB1 mutations were shown to cooperate with KRAS to promote PDAC through suppression of the p21-dependent growth arrest mechanism [47]. Under condition of metabolic stress, LKB1 acts through phosphorylation of AMP-activated protein kinase (AMPK), which is a central metabolic sensor [48]. AMPK is also activated when ATP concentration falls and 5’AMP concentration rises in response to nutrient deprivation or hypoxia, increased intracellular Ca2+ and/or drug (e.g. metformin) administration [49]. Activated AMPK is well known to inhibit mTOR signalling [49].
EGF/EGFR signalling pathway
The EGFR overexpression is observed in 30-90% of pancreatic cancer cases [50] Overexpression of EGFR probably occurs at early stages in PDAC by both genetic rearrangement and gene amplification mechanisms [50] EGFR family members induce cell proliferation, angiogenesis, motility, invasion, metastasis, survival and epithelial-mesenchymal transition (EMT) and reduce apoptosis through activating the downstream signalling pathways, including KRas/B-raf-MEK/ERK, JAK/STAT, PI3K/AKT/mTOR and Ca2+⁄CaM signalling [50,51] Increased co-expression of EGFR and its ligand in pancreatic cancer was associated with more liver metastasis and poorer prognosis [52].
TGFB/SMAD signalling pathway
Individual components of Transforming growth factor beta (TGFB) signalling pathway are deregulated in PDAC, including inactivation of TGFBR2 and SMAD4/Deleted in pancreatic carcinoma, locus 4 (DPC4) genes and overexpression of TGFB [53,54]. TGFB signalling has been implicated in cancer cell proliferation, differentiation, invasion, tumour angiogenesis, extracellular matrix deposition and suppression of anti-tumour immunity [53]. Inactivation of SMAD4 tumour suppressor gene is found in around 60% of PDACs, especially in high-grade PanIN-3 [53,54]. Loss of SMAD4 leads to the development of widespread metastasis in PDAC and decreased survival [54].
Cell cycle control and DNA damage-response pathways
Mutations in genes controlling cell cycle and DNA damage response have been implicated in PDAC. These mutations can be inherited from parents, or can be acquired by carcinogens such as cigarette smoke carcinogens, or by chance. Deficient DNA damage response and cell cycle checkpoints lead to accumulation of mutations, genomic instability and uncontrolled proliferation.
Cyclin-dependent kinase inhibitor 2A (CDKN2A) is inactivated in ~95 % of PDACs with the vast majority of alteration arising as early as the PanIN-2 stage [18,54]. CDKN2A has several alternative splicing sites that generate transcript variants including cyclin-dependent kinase inhibitor, “p16 (p16INK4a)” and p53-activator “alternate open frame” (ARF, p14ARF ) [55]. p16 inhibits phosphorylation of retinoblastoma (RB), thereby blocking entry into the S (DNA synthesis) phase of the cell cycle [55]. Loss of p16 therefore leads to uncontrolled G1/S transition and unregulated cell division.
Inactivating mutations of TP53 have been detected in high-grade (PanIN-3) primary PDACs and metastatic lesions in >50 % of cases [18,55]. TP53 is involved in cell cycle arrest, apoptosis, senescence, DNA repair and metabolism to maintain genomic stability [55]. Upon stress, particularly under the genotoxic stress, p53 is activated and stabilized by action of both Ataxia Telangiectasia Mutated (ATM) and p14ARF [55]. In addition to p53, ATM activates several other key proteins such as BRCA1, fanconi anemia group D2 protein (FANCD2), and serine/threonine-protein kinase CHK2 to initiate activation of the DNA damage checkpoint, leading to cell cycle arrest, DNA repair or apoptosis [55]. Loss of both ATM and p14ARF function have been well documented in PDAC [11,55]. Mutant TP53, which is unable to bind DNA, is incapable of stimulating the production of the p21 tumour suppressor protein, such loss of p21 expression having been detected in 30-60% of PDAC cases [47,56].
Germline mutations in BRCA1 and BRCA2 have been reported in familial cases of PDAC [12,57]. Proper function of BRCA1/2 is required to form a complex with a repair protein RAD51 and a partner called “partner and localizer of BRCA1/2” (PALB2) [58,59] This complex coordinate homologous recombination (HR) comprised of a series of interrelated pathways that function in the repair of DNA double-stranded breaks (DSBs). In the presence of BRCA mutations, if base-excision repair (BER) rescue pathway, regulated by Poly(ADP-ribose) polymerase (PARP) enzyme, is not affected, it maintains genomic stability [60]. Defects in these pathways lead to an accumulation of DNA damage, genomic instability, radioresistant DNA synthesis, impaired cytokinesis, proliferation arrest, hypersensitivity to DNA damaging agents and cell death [57-60].
Germline mutations in Mismatch repair (MMR) have been reported in familial cases of PDAC [12]. MMR genes are highly conserved biologically and play a key role in maintaining genomic stability [12,61]. Defects in MMR functions are associated with genome-wide instability, resistance to chemotherapeutics agents and abnormalities in meiosis all of which can contribute to aggressive tumour phenotypes including early-onset PDAC [11,61].
Acidic microenvironment is a major feature of tumour tissue that promotes aggressive phenotype. It is well described that in PDAC, both oncogenic KRas signalling and hypoxia increases the “glycolytic switch” that results in increased production and export of lactate, attributing to formation of acidic microenvironment [31,32]. In addition to lactate, an excess amount of CO2 may be produced through the pentose phosphate pathway in tumour cells and can be an alternative cause of a lower extracellular pH. Since Carbonic anhydrase 9 (CA 9) is overexpressed in hyperplastic ductal epithelium and PDAC, it catalyses the reversible hydration of carbon dioxide to bicarbonate and protons (CO2 + H2O ↔ HCO3- + H+) [62]. This reaction takes place in the extracellular domain of the enzyme, where bicarbonate is shuttled into the cytoplasm through specific transporters to buffer intracellular pH, while H+ remains in the extracellular space lowering extracellular pH [62]. Thus, CA 9 helps to produce and maintain an alkaline intracellular pH favourable for tumour growth and survival [63]. Meanwhile, CA 9 participates in the generation of an increasingly acidic extracellular space, facilitating cell invasion [64] Other mediators of increased acid extrusion in PDAC cells include Na+/H+ exchangers (e.g., NHE1), various HCO3− transporters (e.g., sodium bicarbonate cotransporter 4/SLC4/NBC), H+ pumps (e.g., V-type H+-ATPases) and lactate-H+ cotransporters (e.g., monocarboxylate transporters (MCTs)) which are upregulated in PDAC (Figure 4) [65]. In particular, the EGF/KRas/NHE1 pathway is implicated in the early progression of PDAC by localized extracellular acidification and induction of an aerobic glycolytic phenotype with higher metastatic potential [66]. Other families of ion channels that have proton conductivity have been also implicated in the pathogenesis of PDAC. Among them are TRP cationic channel of the ‘melastatin-related’ type (TRPM), type 8 (TRPM8), type 7 (TRPM7) and the Transient receptor potential canonical isoform 1(TRPC1) channel, which play roles in proliferation migration, invasion and metastasis [67,68].
Dysfunction of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) also leads to acidification within the acinar lumen. CFTR functions as an anion transporter and facilitates ductal HCO3- secretion [69]. Mutation in CFTR leads to faulty Cl- re-circulation and, HCO3- secretion, reducing pH within the acinar lumen, inhibiting acinar endocytosis of secretory granule protein and reducing the solubility of secreted luminal protein within the acinar lumen. This blocks ducts by mucus and digestive enzymes, followed by destruction of acini, inflammation, and fibrosis [69]. Thus, one or more of these factors may contribute to the development of acute and chronic pancreatitis, and PDAC [12]. Hence, it is therefore not surprising that heterozygous mutations in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene are associated with pancreatic insufficiency, CP, familial cases of PDAC and early-onset PDAC [12,69].
Epigenetic alterations contribute to the development of PDAC. The main epigenetic mechanisms that may affect gene expression include DNA methylation, histone modification, and micro-RNA expression. Alteration in gene expression patterns can cause the activation of oncogenic pathways and the silencing of tumour suppressor and activation of oncogenes leading to the neoplastic changes. Not surprisingly, epigenetic deregulations that occurs from PanIN lesions to invasive PDAC affects virtually all cell functions such as cell-cycle control (e.g., p16), DNA-damage response (e.g., MLH1 (human mutL homolog 1)), proliferation (e.g., RUNX3 (Runt related Transcription Factor 3), evading apoptosis (e.g., RPRM (reprimo)), sustained angiogenesis (e.g., miR-34a), migration and invasion (e.g., S100 Calcium Binding -Protein A4 (S100A4)) [70-72]. The currently available literature on epigenetic alterations in PDAC are summarised in Table 3.
Table 3. Overview of some frequent epigenetic alterations involved in the pathogenesis of PDAC [70-72].
Epigenetic alterations |
Gene affected |
Known or predicted function |
DNA hypermethylation |
CDKN2A |
Cell-cycle control |
|
CCND2 |
Cell-cycle control |
|
MLH1 |
DNA-damage response |
|
RPRM |
P53-induced cell cycle arrest |
|
BNIP3 |
Hypoxia-induced cell death |
|
RASSF1 |
Inhibitor of cell growth |
|
RUNX3 |
Regulation of proliferation and apoptosis |
|
ZEB2 |
Regulator of growth and development |
|
UCHL1 |
Regulation of proliferation and differentiation |
|
SPARC |
Cell cycle progression inhibition, cell matrix-interaction |
|
MIR148A |
Proliferation, colony formation |
|
CDH1 |
Cell-cell contact |
|
CLDN5 |
Cell-cell contact |
|
SFRP1 |
Madulator of Wnt signalling |
|
NPTX2 |
Neuronal transport |
|
PENK |
Neuropeptide precursor |
|
ppENK |
Neutopeptide transmitter |
|
|
|
DNA hypermethylation |
S100P |
Cell cycle progression and differentiation |
|
LCN2 |
Epithelial differentiation |
|
MIR200 |
EMT |
|
MSLN |
Cell surface antigen/cell adhesion |
|
CLDN4 |
Cell adhesion/invasion |
|
PSCA |
Cell surface antigen/cell differentiation |
|
S100A4 |
Motility, invasion, tubulin polymerisation |
|
SERPINB5 |
Regulation of cell motility and cell death |
|
TFF2 |
Secretory polypeptide/epithelial repair |
miRNAs |
Expression level |
Target gene |
Impact on cell function |
Oncogenic miRs |
↑miR-21 |
PTEN |
↑proliferation,invasion, chemoresistance |
|
↑miR-221 |
CDKN1B |
↑cell cycle progression, chemosensitivity |
|
↑miR-10a |
HOXA1, HOXB1, 3 |
↑invasion and metastasis |
|
↑miR-224 |
CD40 |
↑invasion, metastasis |
|
↑miR-155 |
TP53INP1 |
↓apoptosis |
|
|
|
|
Tumour-suppressive miRs |
↓Let-7 |
KRAS |
↑proliferation |
|
↓miR-421 |
Smad4 |
↑proliferation, colony formation |
|
↓miR-34a |
TP53 |
↓apoptosis and DNA repair, ↑cell cycle progression and angiogenesis |
|
↓miR-34 |
Bcl-2, Notch |
↑proliferation, ↓apoptosis, ↑invasion, |
|
↓miR-143 |
GET1, GET2, KRAS |
↑proliferation, invasion, migration |
|
↓miR-146a |
EGFR |
↑invasion |
|
↓miR-200 family |
ZEB1, SIP, EP300 |
↑EMT, metastasis |
(↑, increased; ↓,decreased)
Cancer stem cells (CSCs) are described as phenotypically distinct cancer cells that possess enhanced tumour-initiating potential, self-renewal, and the ability to recapitulate the cellular heterogeneity of the original tumour [73]. Pancreatic cancer stem cells (PanCSCs) represent 0.5% to 1.0% of all pancreatic cancer cells, expressing the surface markers CD44+, CD24+, and epithelial-specific antigen (ESA)+ [73]. The CD44+ CD24+, ESA+ PancSCs show a strong transcriptional upregulation of the Sonic hedgehog (Shh) ligand and the polycomb group (PCG) gene family member BMI-1, controlling cell fate, self-renewal and multi-lineage differentiation [74]. Integration of EGFR and Hedgehog signalling induces expression of SOX2, SOX9, CXCR4, Fibroblast Growth Factor-19 (FGF-19) that are required for tumour-initiation [75]. In addition, MET, Notch, Wnt/catenin beta-1, PI3K/AKT/mTOR, and TGFB signalling pathways are reported as contributors to PanCSCs biology [76,77].
PDAC also contains 1% to 3% of CD133+ cancer cells, that are highly resistant to chemotherapy and partially overlap with CD44+ CD24+, ESA+ PancSCs [78]. Some of CD133+ cancer cells also show high expression of CXC chemokine receptor (CXCR4), a receptor for stromal-derived factor 1 (SDF 1/CXCL12) [78]. Importantly, the invasive front of human pancreatic cancer tissue specimens from patients with more advanced metastatic disease express high level of CXCR4+, indicating the role of CD133+ and CXCR4+ cells in metastasis [78]. Accordingly, blocking CD133+/CXCR4+ cells prevented metastasis of tumour xenograft in mice [78]. SDF 1 is strongly expressed in lung, liver, bone marrow, and lymph nodes, sites that are commonly affected by pancreatic cancer metastases [79]. Hypoxic microenvironment also potentiates PanCSCs to acquire migratory ability by inducing EMT signalling and expression of CA 9 [80]. Indeed, hypoxia enhanced clonogenic survival and migration of PanCSCs through upregulating expression of autophagy-related genes [81]. PanCSCs cells rely on less glycolytic and more dependent on mitochondrial respiration for energy production compared to mutant KRas-expressing pancreatic cancer cells, consequently, they generate more reactive oxygen species (ROS) [82]. Thus, upregulation of autophagy confers protection and resistance against intrinsic and extrinsic stressors such as ROS, nutrient deprivation and hypoxia.
Desmoplasia constitutes a dynamic compartment of PDAC that is critically involved in tumour formation, progression and metastasis, and may even be responsible for the initiation of oncogenesis in the presence of normal epithelial physiology. Reciprocal interaction between cancer cells and stromal cells modulate the production and composition of extracellular matrix (ECM), and increase the recruitment of inflammatory cells and promote the proliferation and activation of pancreatic cancer stellate (Figure 4).
In PDAC, the immune reaction consists of largely immunosuppressive and pro-tumourigenic elements that exist even in the early stages [83,84]. In vivo lineage tracing experiments demonstrated that paracrine interactions between inflammatory cells and cells possessing stem cell properties induce EMT and metastasis to liver, this process occurring even before the carcinoma becomes detectable by standard histology [85]. While both clinical and animal models provide strong evidence for inflammatory stroma initiating PDAC development and allowing progression, [83,84] there are many lines of evidence supporting the view that normal pancreatic stroma suppresses pancreatic tumour formation [86]. For instance, human stromal cells derived from adipose tissue strongly inhibit PDAC proliferation both in vitro and in vivo and induce tumour cell death [87].
It is well demonstrated that long-term progressive inflammatory conditions caused by obesity, genetic factors (e.g. mutations in PRSS1 or SPINK1), life-style factors (e.g., alcohol and tobacco use) or other tumour associated factors (e.g. mutations in KRAS), are able to potentiate pancreatic neoplasia [32,87,88].
A study investigating the contribution of obesity to pancreatic carcinogenesis revealed that a high-fat diet activates KRas signalling via COX-2, leading to pancreatic inflammation and fibrosis, and subsequent development of PanINs and PDAC [30]. Indeed, tobacco-related carcinogens, including nitrosamines, polycyclic aromatic hydrocarbons and their metabolites, cause mutations in KRAS and TP53 genes, and promote pancreatic inflammation and PDAC [20]. Oncogenic KRas signalling a pro-tumourigenic microenvironment through the upregulation of pro-inflammatory cytokines, such as interleukin (IL)-6, IL-11, tumour necrosis factor (TNF)-alpha, IL-1α, by PanIN cells [88]. These cytokines induce proliferation and survival of PanIN cells through activation of the JAK2/STAT3 and NF-κB pathways in an autocrine manner and recruit immune (particularly myeloid) cells [88]. Recent studies have identified a B-cell subpopulation presented in PanINs, promoting the pro-tumorigenic (TH2-type) macrophage phenotype (tumour-associated macrophages-TAM-) leading to immune suppression and PDAC progression [89]. Recruited immune cells secret immunomodulatory mediators and growth factors (e.g., IL-35, IL-6, IL-11, TNF-alpha, IL-1alpha, IL-10, IL-1beta, IL-2, ROS, EGF, TGFB, HH and MMPs) to create a positive-feedback loop and to suppresses cytotoxic T cell response (CTL) [88-90]. These cytokines promote EMT, proliferation and survival of PanIN and PDAC cells and inhibit oncogene-induced senescence [87,88]. Importantly, TNF-alpha stimulates ROS accumulation in epithelial cells, causing DNA damage and genomic instability thereby promote oncogenic mutagenesis [30,91]. In addition, cytokines activate Notch, and Hedgehog signalling synergistically with KRas to accelerate PDAC development [30,95]. Myeloid derived suppressor cells (MDSCs), immunosuppressive cell type, suppress CTL response and induce development of regulatory T cells (Tregs) [84,91,92]. The majority of the T-lymphocytes in PDAC are Tregs, involved in suppression of the immune response, and significantly increased in the blood of PDAC patients as well as in the pancreatic tissue [92,93]. Accumulation of Tregs and MDSCs positively correlate with the progression of disease and negatively correlate with patient survival [92]. Notably, tumour-derived lactate production increases number of MDSCs that inhibit Natural Killer (NK) cytotoxicity [94]. PDAC cells also express several factors such as granulocyte macrophage colony-stimulating factor (GM-CSF), IL-10,-4,-6, TGFB, that in turn suppress dendritic cell (DC) maturation, so limiting T-lymphocyte proliferation [93,95]. Accordingly, decreased circulating DCs and decreased NK activity are observed in PDAC patients [96]. Indeed, PDAC cells can induce apoptosis of infiltrating T cells by secretion of Fas ligand as well as by downregulating expression of human leukocyte antigen (HLA) I molecules and Fas signalling, thus blocking and evading the immune response at the tumour site [97,98]. In fact, PDAC cells express a variety of cancer-associated antigens that can potentially be recognised by T lymphocytes [97,99,100]. Several studies have revealed that tumour-specific Cytotoxic T Lymphocytes (CTL/CD8+ T) precursors present in peripheral blood and bone marrow of pancreatic cancer patients [99,100] Indeed, the infiltration of the tumour by effector CD8+, CD4+ T cells and dendritic cells was found to be a good indicator of the patient’s outcome after surgical treatment [100].
Pancreatic stellate cells (PSCs) (also known as myofibroblasts or cancer-associated fibroblasts) are predominant mesenchymal type cell within the PDAC stroma [101,102]. In the normal, healthy pancreas, they are found in small numbers in their quiescent state and located in the periacinar and periductal regions of the exocrine pancreas [101]. They have characteristic retinoid-containing fat droplets in their cytoplasm, a low mitotic index and a low capacity for ECM synthesis [101] PSCs are activated by a range of factors including proinflammatory cytokines, growth factors, oxidative stress, toxins (e.g.,alcohol and its metabolites, endotoxins), hypoxia, increased interstitial pressure, a high-fat diet and hyperglycaemia [30,102]. Upon activation they transform from a quiescent state to the activated-myofibroblastic state [101,102]. Activated PSCs loss fat droplets (containing retinoic acid), express alpha smooth muscle actin (α-SMA) synthesize growth factors (e.g.,TGFB, VEGF, HGF, TNF-alpha, PDGFB) and inflammatory cytokines (such as IL-6, IL-1beta) as well as excessive amounts of ECM protein (including collagen, laminin, fibronectin, and periostin) which form the fibrous tissue [16,103-105]. Once activated, PSCs can preserve their own activity by forming autonomous feedback loops, as well as promoting proliferation, migration invasion, metastasis, EMT and survival of PDAC cells [16,103-105]. Reciprocally, tumour cells produce growth factors to induce PSCs cells to secrete ECM protein [105]. PSCs also regulate the re-absorption and turnover of the stroma, mainly through the production of MMPs [102]. Increased expression of periostin and collagen, the main products of PSCs, were detected in the stroma of PanIN, IPMNs and PDAC and its expression increases in parallel with the stages of malignant transformation [15,104-106].
PSCs have an important role in mediating the immunosuppressive microenvironment in PDAC by promoting proliferation and activation of MDSCs via secreting cytokines and VEGF, impairing the survival of T cells, recruiting Treg and sequestering CTLs via CXCL12/CXCR2, and CXCL12/CXCR4, respectively, impeding their contact with tumour cells [107-109]. Therefore, T-lymphocytes were shown to surround the pancreatic lesions and found more frequently in the fibrotic interstitial tissue than in the intraepithelial area of the PDAC [109]. PSCs release IL-33 to activate mast cells to produce pro-inflammatory cytokine, MMPs production promoting PDAC progression [91]. In addition, SDF 1 secretion by PSCs cells induce invasion of cancer cells through activating SDF 1/CXCR4 axis [110]. These gradients of SDF 1 may attract PSCs and PDAC cells and regulate proliferation and invasion at specific metastatic sites [110]. Indeed, PSCs accompany cancer cells to distant metastatic sites where they may facilitate the seeding, survival and proliferation of cancer cells [111]. Interestingly, Tien et al demonstrated that PDAC cells stimulate activation of hepatic stellate cells via PDGF, FGF2, TGFB to modify the liver stroma to become more suitable for their survival [112] Given the similarities between HSCs and PSCs and similar collagen distribution patterns between primary pancreatic tumours and related secondary liver metastases, it is reasonable to speculate that HSCs play a critical role in the metastasis of PDAC cells to the liver [113].
Recent studies have also implicated PSCs in neural growth and perineural invasion (PNI) in PDAC.17 A positive correlation between the extent of desmoplasia and the degree of neural invasion in human PDAC has been reported [17]. It is well established that PNI by inflammatory, immune or cancer cells damages nerves and gives rise to the classically prognostic pancreatic neuropathic pain associated with PDAC [17] Perineural invasion appears, based on studies of in vivo models, to be triggered via the Sonic hedgehog (Shh) signalling pathway which in turn activates PSCs with altered, mutant gene expression profiles in the tumour microenvironment and leads to tumour progression [114]. Neuron growth and elongation are also influenced by collagen, fibronectin and hyaluronic acid which are predominantly produced by PSCs [115]. In addition glial-derived NGF, cholinergic and sympathetic inputs promotes cancer cell invasion and proliferation [116,117].
A dense collagen-rich fibrotic ECM is one of the hallmarks of the PDAC. Despite the high metastatic potential of PDAC, it seems that this dense fibrotic structure may serve as a barrier to migration and invasion. In a paradoxical twist, the desmoplastic reaction functions in such a way as to stimulate PDAC progression and metastasis.
Besides being rich in immunomodulatory mediators and growth factors, ECM contains multiple cell-matrix interaction modulators, including thrombospondin, periostin, tenascin C (TNC), secreted protein acidic and rich in cysteine (SPARC), vitronectin, biglycan, collagens (predominantly type I, III, and IV), laminin and fibronectin as well as proteoglycans and glycosaminoglycans [118]. Integrin and CD44 signalling are among the important means of cellular communication with the ECM in PDAC. Multiple integrin subunits which can interact in a variety of combinations to form unique receptors with differing affinities for the ECM protein promote adhesion, survival, growth, migration, and invasion [119].
In addition to composition, the stiffness of ECM regulates biology of tumour [119]. A positive feedback loop was described between collagen I, MT1-MMP and TGFB signalling promoting the establishment and maintenance of the desmoplastic reaction and supporting migration [119] Notably, increased expression of collagen was used to calculate an index for the activation of the stroma in each tumour and the higher this index was found to be positively correlated with the worse prognosis for patients with PDAC [15,16].
In addition to providing signalling scaffolds, sustained fibrogenesis together with fluid-trapping mucopolysaccharides (e.g. hyaluronan) act as a barrier to perfusion causing high interstitial fluid and changes the organisation and structure of vessels and microcapilllaries [120]. All these modifications alter vascular permeability to nutrient and therapeutic and cause hypoxia [110]. Hypoxia induce stromal cells and PDAC cells to release angiogenic factors (e.g., VEGF, FGF, angiopoietin 2, periostatin, COX-2, neuropilin-1) inducing angiogenesis and causing tumour overgrowth. Activation of angiogenic signalling pathways have been found to correlate with poor prognosis in PDAC patients and is also associated with liver and lung metastasis [121-123]. Meanwhile, continuous activation of PSCs cause excessive deposition of ECM molecules and induce PDAC cells to produce endostatin, an inhibitor of endothelial proliferation and potently inhibits angiogenesis [124]. Therefore, in contrast to expectations, such manipulation of the microenvironment overwhelms local pro-angiogenic properties creating hypovascular microenvironment and a cirrhotic/hypoxic tissue [124]. These findings may provide explanation to insufficiency of anti-angiogenic therapies in PDAC and suggest novel therapeutic approaches targeting cancer-stroma interactions.
Diagnosis
PDAC usually presents clinical symptoms late in the course of the disease when the tumour is already advanced or has already spread beyond the pancreas or metastasized to other organs. The presenting symptoms of PDAC depend on the location of the tumour within the pancreas, as well as on the stage of the disease. However, most symptoms are vague and could be attributed to many different conditions, leading to late detection of malignancies. Another contributing factor to late detection is that the functionality of the pancreas is relatively unaffected until over 50% of its tissue is rendered non-functional. Furthermore, the position of the pancreas deep in the abdomen makes it inaccessible for physical examination by primary care physicians. Thus, the accurate and early detection of PDAC is extremely difficult with currently available diagnostic means.
The majority of the PDACs are located in the head of the pancreas. Although this does not necessarily alter the biology of the disease, patients with tumours in the body or tail of the pancreas have an anatomical advantage over those with tumours in the pancreas head, because they are less susceptible to biliary obstruction and, therefore, less likely to require interventions that could increase their risk of infection, especially when on treatment with chemotherapy.
Clinical history
Common clinical features include abdominal persistent pain, particularly epigastric pain that radiates to the back, unexplained weight loss, jaundice, clay-coloured stools, dark urine, nausea and/or vomiting, steatorrhoea, malaise, and coagulopathy [125]. PDAC originating anywhere in the pancreas can be associated with new onset or worsening of existing diabetes [131]. Diabetes mellitus is present in around 70% of patients, usually with a diabetes history less than 2 years [126]. Later symptoms are related liver metastasis and/or invasion of adjacent organs (stomach, colon) or of the peritoneal cavity, which may lead to ascites [126]. Jaundice and liver function abnormalities may also indicate that cancer has metastasised to the liver [126] Occasionally, patients present with acute pancreatitis, migratory thrombophlebitis, or hypercalcaemia [3]Depression is also common in patients with pancreatic cancer [127].
Laboratory examination
The laboratory findings in PDAC patients are usually nonspecific. However, initial blood work generally include a complete blood count (CBC), complete metabolic panel (CMP), serum amylase and/or lipase, and tumour markers (Cancer antigen 19-9 (CA 19-9), Du-Pan 2, carcinoembryonic antigen (CEA), Span-1) [126]. Total and direct bilirubin measurements and liver-function tests including serum aminotransferases (AST/ALT) and alkaline phosphatases may reveal evidence of biliary obstruction, and liver metastasis [126]. Serum amylase and lipase levels may be elevated from pancreatic ductal obstruction or pancreatic tissue injury [128]. CA19-9 is the only tumour marker that is approved by the US Food and Drug Administration for the use of pancreatic cancer [129]. However, CA19-9 is not a specific tumour marker for PDAC so should not be used independently for PDAC screening as levels may be elevated in other conditions such as pancreatitis, gallstones, cholestasis, liver disease and various inflammatory diseases [119]. In addition, the test is ineffective in individuals with no functional Le enzyme, which plays a role in post-translational modification of CA 19-9 [129].
Radiology
Currently, there is no single method that provides sufficient sensitivity and specificity by itself, therefore, combinations of different imaging modalities and blood tests are employed in the preoperative diagnosis and staging of patients with suspected PDAC. Ultrasonography (US), computed tomography (CT), magnetic resonance imaging (MRI), positron emission tomography (PET), endoscopic ultrasonography (EUS), endoscopic retrograde cholangiopancreatography (ERCP), magnetic resonance cholangiopancreatography (MRCP), and multi-detector row computed tomography (MDCT) are currently available pancreatic imaging techniques that are used in characterization of pancreatic focal lesions, initial staging, surgical and therapeutic planning, and assessment of the treatment response [126,130]. MDCT preferably complemented by EUS is more sensitive for the early detection of lesions, and allows relatively easy access to the pancreas for tissue diagnosis using fine-needle aspiration (FNA), as well as providing further important information for use in tumour staging [130].
Staging
Physical examination, imaging tests, laboratory tests, pathology reports and surgical reports are used to accurately stage disease. Once a mass is identified and FNA confirms the diagnosis, EUS can determine tumour size, extent of lymph node metastases, and assess for portal venous system involvement to complete staging. Currently, staging systems are used to predict patient outcome or suggest best treatment options depending on the stage of the disease. Several staging systems or consensus statements have been developed by different societies or academic practices to stage patients with PDAC. These include those developed by the American Joint Committee on Cancer (AJCC),131 the National Comprehensive Cancer Network,132 the American Hepato-Pancreato-Biliary Association, the Society of Surgical Oncology, the Society for Surgery of Alimentary Tract and the University of Texas MD Anderson Cancer Center.133 All of these staging systems primarily depend on tumour size, location within the pancreas, extension beyond the pancreas with or without contact with the adjacent vessels, and the presence or absence of metastatic lesions [131-133]. The major areas of spread are lymphatic channels and regional lymph nodes, retropancreatic tissue connection, liver, peritoneum, bone marrow, lung and local invasion of major vascular structures, especially the portal and mesenteric venus tree, as well as the estrapancreatic nerve plexus [134]. According to the AJCC tumour-node-metastasis (TNM) classification, which is based on assessment of resectability by means of helical CT, T1, T2, and T3 tumours are potentially resectable, whereas T4 tumours, which involve the superior mesenteric artery or coeliac axis, are unresectable (Table 4). Tumours involving the superior mesenteric veins, portal vein, or splenic veins are classified as T3, because these veins can be resected and reconstructed [134]. A multidisciplinary team with expertise in surgery, diagnostic imaging, pathology, interventional endoscopy, and medicinal and radiation oncology is strongly recommended to determine which patients are eligible for surgery with adjuvant therapy.
Table 4. AJCC 7th TNM classification of tumours of the exocrine pancreas
TNM classification |
|
Primary Tumour (T) |
TX: Primary tumour cannot be assessed |
T0: No evidence of primary tumour |
Tis:Carcinoma in situ |
T1: Tumour limited to the pancreas, 2cm or less in greatest dimension |
T2: Tumour limited to the pancreas, more than 2 cm in greatest diameter |
T3: Tumour extends beyond pancreas but no involvement of celiac axis or superior mesenteric artery |
T4: Tumour involves the celiac axis or the superior mesenteric artery (unresectable) |
|
Regional Lymph Nodes (N) |
MX: Regional lymph nodes cannot assessed |
N0: No regional lymph node metastasis |
N1: Regional lymph node metastasis |
|
Distant Metastasis (M) |
MX: Distant metastasis cannot be assessed |
M0: No distant metastasis |
M1: Distant metastasis |
|
Staging group |
Stage 0 |
Tis |
N0 |
M0 |
Localized within pancreas, resectable |
Stage 1A |
T1 |
N0 |
M0 |
Localized within pancreas, resectable |
Stage 1B |
T2 |
N0 |
M0 |
Localized within pancreas, resectable |
Stage IIA |
T3 |
N0 |
M0 |
Locally invasive, resectable |
Stage IIB |
T1, 2, or 3 |
N1 |
M0 |
Locally invasive, resectable |
Stage III |
T4 |
Any N |
M0 |
Locally advanced, unresectable |
Stage IV |
Any T |
Any N |
M1 |
Distant metastases |
Resec table PDAC
Currently the only accepted potentially curative modality is complete surgical resection with negative surgical margins for patients with localised resectable PDAC. An expert consensus has developed criteria to define tumour resectability, thereby patients who will benefit from surgery [132,135,136]. The most common type of a surgery method used for resection is known as the Whipple procedure in which the tumour-bearing region of the pancreas along with a portion of stomach, duodenum, gallbladder, and part of bile duct are removed and the remaining regions are reattached to support digestive capabilities of the patient [132].
Surgical therapy for PDAC is limited; thus, post-operative therapy (adjuvant therapy) is considered the standard of care for patients with resectable PDAC. Adjuvant treatment with both chemotherapy and radiation therapy were demonstrated improvements in disease-free survival and overall survival (OS) rates [137]. Currently there is no universal consensus on the adjuvant treatment of surgically resectable PDAC. Based on results from the six-prominent adjuvant prospective randomized phase III trials (GITSG [138] EORTC [139] ESPAC‐1 [140,141] CONKO‐001 [142] ESPAC‐3 [143] and RTOG 97‐04 [144]) for PDAC, chemotherapy with gemcitabine or 5-fluorouracil (5-FU) or 5-FU plus leucovorin (folinic acid) for six months without or with radiation represents a standard care. The median disease-free survival following complete resection of PDAC and gemcitabine administration is 13.4 months and 6.7 months for untreated patients [143]. Gemcitabine alone is often recommended as the current standard adjuvant chemotherapy because less adverse effects were observed with gemcitabine compared to 5-FU treatment, while no significant differences in survival rates between the two treatment arms (median survival from resection was 23 and 23.6 months, respectively) were found [143].
Patients with borderline resectable tumours may become surgically resectable after appropriate preoperative (neoadjuvant therapy) chemotherapy or chemoradiotherapy [145,146]. In a large report of borderline resectable PDAC, the preoperative gemcitabine-based regimens have been associated with low rates of microscopically positive margins on resection, greater treatment effect, low rates of postoperative local recurrence, and improved OS in those undergoing resection compared with 5-FU- or paclitaxel-based regimens [145,146]. Indeed, the survival results were found comparable with those reported in adjuvant therapy trials [146]. While median OS still does not exceed 2 years, 5-year survival range from 40-50% and 15-20%, respectively [146]. This neoadjuvant approach allows for identification of the subset of patients that was most likely to benefit from resection as evidenced by favourable median survival in this group [145,146].
Non-resec table PDAC
In the vast majority of cases, PDAC patients are diagnosed with locally advanced, inoperable tumours (~40% of cases) or metastatic disease (~40-45% of cases) and median survival reported for these groups are with median survival 8-12 months and 3-6 months, respectively [136,147]. The initial metastatic site is important for prognosis of patients with metastatic PDAC, while patients with extrahepatic metastasis had better prognosis than those with hepatic metastasis, patients with both had the worst prognosis [148]. Locally advanced PDAC patients with no evidence of distant metastasis are defined as surgically unresectable, and the goal of the therapy, as in metastatic disease, is prolongation of survival, symptom palliation and disease control [149]. Systemic chemotherapy offers benefit for advanced PDAC, improving symptoms and OS when compared to best supportive of care. In 1997, Burris et al.[150] reported that gemcitabine was superior over 5-FU in patients with advanced disease. In this phase III study, the researchers demonstrated that the median OS durations were 5.65 and 4.41 months for gemcitabine-treated and 5-FU-treated patients (P = 0.0025) and the 1-year survival rate was 18% and 2% for the gemcitabine and 5-FU group, respectively [150]. Since the first demonstration of clinical benefit/efficacy and safety profile in 1997, gemcitabine has been the cornerstone of first-line treatment worldwide for patient with borderline, locally advanced and metastatic PDAC [150]. Since then, many phase II trials have reported promising activity for various cytotoxic (capacitabine, cisplatin, irinotecan, oxaliplatin, pemetrexed, exatecan) and targeted agents (cetuximab, tipifarnib, sorafenib, axitinib, bevacizumab) in combination with gemcitabine. However, phase III trials of these combinations did not result in meaningful clinical improvement or survival benefit in advanced PDAC patients [147]. One phase III study showed that the addition of erlotinib to gemcitabine improved OS compared with gemcitabine alone, but this benefit was small (6.2 vs 5.9 months) and accompanied by a substantial increase in toxicity [151].
In 2011, the PRODIGE 4 trial demonstrated that, the cytotoxic combination regimen FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan, oxaliplatin) significantly improved median OS to 11.1 months compared to 6.8 months for gemcitabine treatment [152]. However, treatment with FOLFIRINOX resulted in significantly more adverse events of >grade 3 in terms of neutropenia (45.7% for FOLFIRINOX vs. 21.0% for gemcitabine), febrile neutropenia (5.4% vs. 1.2%), thrombocytopenia (9.1% vs. 3.6%), diarrhoea (12.7% vs. 1.8%) and peripheral neuropathy (9.0% vs. 0%), while the incidence of elevation of alanin aminotransferase was decreased (7.3% vs. 20.8%) [152]. Currently FOLFIRINOX treatment is limited to patients with good performance status and the vast majority of PDAC patients still receive gemcitabine, which is recommended as a monotherapy because it provides clinical benefits by relieving symptoms and prolonging survival (typically by two to three months) [152,153]. To improve tolerability and reduce the risk of serious toxicity, dose modification studies suggest that dose attenuation of bolus 5-FU as well as irinotecan improve tolerability without compromising efficacy in locally advanced and metastatic PDAC patients [154,155]. Another study used a modified FOLFIRINOX regimen for patients with advanced non-metastatic PDAC and showed fewer haematologic toxicities and maintained an impressive resection rate in the neoadjuvant setting [156].
In 2012, the MPACT trial in advanced PDAC demonstrated that nab-paclitaxel in combination with gemcitabine improved the response rate (7% in gemcitabine alone vs 23% in combination), progression-free survival (PFS) (from 3.7 months to 5.5 months), and OS from 6.7 months to 8.5 months, compared to single agent gemcitabine [157,158]. As expected, the combination arm increased the haematological toxicity profile, and non-haematologic clinical toxicity such as neuropathy, fatigue, alopecia and mucositis, as compared with the gemcitabine arm [157,158]. The toxicity profile for both nab-paclitaxel plus gemcitabine and FOLFIRINOX is similar. However, haematological toxicities and growth factor usage was higher in the FOLFIRINOX regimen, whereas nab-paclitaxel plus gemcitabine demonstrated higher rates of neuropthy [158]. Compared to FOLFIRINOX treatment the overall nab-paclitaxel plus gemcitabine treatment was found to be well tolerated and manageable in most advanced PDAC patients [158].
One of the most important clinical consequences of first-line regimens with FOLFIRINOX or nab-paclitaxel plus gemcitabine is that an increasing proportion of patients experience prolonged disease control, which enables them to receive second-line and maintenance therapies with clinical benefit. Although, today, there is no accepted active regimen for second-line treatment, there are available data from clinical trials of second-line systemic therapy in patients with unresectable PDAC. For instance, a randomized phase III study provides evidence that second-line chemotherapy with oxaliplatin and 5-FU is a good option after first-line therapy with gemcitabine [159]. Additionally, studies have shown that locally advanced and metastatic PDAC, which were primarily resistant to FOLFIRINOX treatment, still responded to a combination treatment with nab-paclitaxel and gemcitabine, with a manageable toxicity profile [160-162]. Monotherapy with gemcitabine was also found to be a reasonable second-line option for patients with advanced PDAC after progression on FOLFIRINOX [163]. Recently, nanoliposomal irinotecan in combination with 5-FU and folinic acid was found to extend survival with manageable safety profile with metastatic PDAC who previously received gemcitabine-based therapy. in a global, randomised, open-label, phase III trial in 14 countries (NAPOLI-1) [164]. Median OS in patients assigned nanoliposomal irinotecan plus 5-FU and folinic acid (n=117) was 6.1 months vs 4.2 months with 5-FU and folinic acid (n=149). On the other hand, median OS did not differ between patients assigned nanoliposomal irinotecan monotherapy (n=151) and those allocated 5-FU and folinic acid (4.9 months vs 4.2 months) [164].
Maintenance therapy is a new era in the treatment of advanced PDAC, which enables prolonged disease control, eventually improving OS. Recently, Reni et al.[165] addressed for the first time the role of a maintenance strategy in the management of PDAC. Sunitinib, a potent inhibitor to Raf, VEGFR and c-Kit kinase, was shown to be promising for maintenance therapy [165] In this phase II study, patients with metastatic PDAC, performance status >50%, and no progression after 6 months of chemotherapy, were randomised to observation (arm A) or sunitinib daily (for a median of 91 days) until progression or to a maximum of 6 months (arm B) [165]. Main grade toxicity was thrombocytopenia, neutropenia, hand-foot syndrome and diarrhoea. The study fulfilled its primary endpoint; while PFS for 6 months was 3.6% (95% confidence interval (CI): 0-10.6%) in arm A, it was 22.2% (95% CI: 6.2-38.2%; P<0.01) in arm B. Although differences in OS did not reach statistical significance, the proportion of patients who were alive at two years was tripled in the arm B compared with arm A [165].
A treatment strategy implementing combination therapy using FOLFIRINOX or nab-paclitaxel plus gemcitabine results in superior disease control and both treatment regimens have been quickly adopted into clinical practice. However, their increased toxicities, additional expenditure and a narrowing patient profile that are fit enough to undergo these treatments limits their use [153,158].
Palliative care
Biliary obstruction, gastric outlet obstruction, cancer-related pain, malnutrition, thromboembolic disease, tumour-related pain and depression are the complications of locally advanced and metastatic PDAC that most require palliative intervention. Palliative care for patients with obstructive jaundice includes biliary bypass or biliary stent. Patients with a life expectancy longer than three months can receive an open or laparoscopic gastrojejunostomy with or without jejunosomy tube; an enteral stent can also be an option. However, for patients with a short life expectancy or poor performance status, an enteral stent can be placed [166,167]. Oral pancreatic enzyme replacement is recommended for exocrine pancreatic insufficiency [168]. Low-molecular-weight heparin and warfarin are used to prevent recurrent thromboembolic disease [166]. Severe, intractable pain, occurs in a majority of patient with PDAC. Persistent pain is positively correlated with the tumour size and infiltration of the intra- and extra-pancreatic nerves [169]. Opioid analgesics have been used for chronic pain [170]. Celiac plexus block has been a common procedure to alleviate pain in patients with unresectable PDAC with potentially fewer adverse events than traditional opioid management [171]. Common, side effects include urinary retention, back pain, diarrhoea, and hypotension. Serious complications such as transient or permanent paraplegia are extremely rare [171].
Overall these procedures provide physiological benefits, including improvements in hepatocyte metabolism, protein synthesis, absorption and digestion of fats, and bacterial clearance [166]. In addition, relief from maldigestion, pruritus, oedema and relief of gastric stasis improve the patient’s physical condition and quality of life (QOL).
Concepts of therapy resistance
Numerous chemotherapy regimens, discussed above, have been tested in recent years and most have uniformly failed to improve OS of PDAC patients. PDAC is well recognised as an extremely challenging disease on multiple fronts. Fuelling this paradigm is a set of underlying biological attributes uncommon to other cancers. There are key attributors: i) intra-, inter-tumoral genetic and epigenetic heterogeneity contributing multiple molecular aberrations, ii) cross-talks and feedback mechanisms between signalling pathways and constitutively active molecules, iii) desmoplastic reaction, iii) evasion of the immune system, and iv) PanCSCs- all of which contributes to biologically and clinical aggressiveness of PDAC. In addition, interpretation of clinical trials, and practical and ethical complications around the selection of patients are most likely reasons for the failure of trials.
The intra- and inter-tumoral genetic heterogeneity is one mechanism whereby PDAC acquires increasing aggressiveness and resistance to therapy. According to a molecular profiling study which compared the most significantly differentially expressed genes/pathways between PDAC and control patient samples, there was little to no overlap between genes/pathways indicating inter- individual heterogeneity of PDAC [172]. In addition, a genetic profiling study showed that there was significant heterogeneity both intra-tumoral and between metastases in the same patient [80,172] Importantly, genetic heterogeneity predominantly occurs in early cancer development and persists after cancer dissemination, resulting in ongoing, parallel and event convergent evolution among different metastases [80,172].
An intriguing study addressing PDAC development and progression based on a computational model that incorporated the number of somatic alterations and the relative proliferation rates of cells as they progress from normal to cancerous and finally to metastatic cells, proposed that PDAC has a long latency from initiation to patient death that is in the order of decades [173]. This finding suggests that there is a long latency to development of metastasis and thus a large window of opportunity for early diagnosis and cure [80]. On the other hand, patients with very small or clinically undetectable primary tumours still have a high risk of developing metastasis. Respectively, experimental data suggests that dissemination of preneoplastic cells is an early event in PDAC even before the carcinoma was detectable by standard histology [80]. These findings recall the fact that adding to genetic heterogeneity, the continuously changing microenvironmental signals exert different selection pressure on cancer cells leading to phenotypic and functional heterogeneity. Thus, the remarkable diversity and adaptability of cancer cells hamper curative therapies for PDAC. In addition, acquisition of de novo genetic lesions over the course of therapy and treatment-induced selection of resistant minor subpopulations of cells that are intrinsically insensitive and already present in the original tumour make the resistance problem even more challenging.
Wide inter-patient and intra-tumoural heterogeneity in the delivery and metabolism of gemcitabine have been reported in PDAC [174,175]. Gemcitabine is a prodrug that requires nucleoside transporters, equilibrative nucleoside transporter (ENT1), and a concentrative nucleoside transporter (CNT), to gain access to the intracellular compartment. After entering the cell, it is activated by a subsequent phosphorylation event by deoxycytidine kinase (dCK) and thymidine kinase 2 (TK2). Being a deoxycytidine analogue, activated gemcitabine (triphosphate-gemcitabine (dFdCTP)) blocks DNA synthesis through inhibition of ribonucleotide reductase (RRPM1, RRM2) and incorporation into DNA and RNA, thereby preventing growth and initiating apoptosis [175]. The majority of intracellular gemcitabine is inactivated by deamination by cytidine deaminase (CDA) [174]. Acquired resistance based on gemcitabine transport and metabolism is associated with deficiency in ENT1, CNT1, CNT3, dCK and TK2 and overexpression of RRM1 and RRM2 [174-179]. Increased expression of miR-21 and decreased expression of miR-200 contribute to chemoresistance to gemcitabine as well as increase aggressiveness of cancer cells [180]. Additionally, loss of p53 function, increased expression of anti-apoptotic protein (Bcl family protein), NF-κB and hypoxia-inducible factor 1-alpha and higher activation of SRC kinase, EGFR, STAT3, PI3K/AKT, Notch and MAPK pathway during pancreatic carcinogenesis confers resistance to gemcitabine [178-184]. Similarly, activation of molecular cascades including anti-apoptotic, SRC kinase and EGFR/AKT provides survival benefits to PDAC cells during the course of prolonged 5-FU treatment [180,186]. 5-FU which is an active form of orally administrated capecitabine, inhibits thymidylate synthase (TS) and incorporate its metabolites into RNA and DNA. Not surprisingly, deficiency in 5-FU transporters (e.g. human concentrative nucleoside transporter (CNT1)) and upregulation of multiple drug resistance-transporters (MRPs) over the course of 5-FU treatment reduces the accumulation and cytotoxicity of 5-FU [179,187,188]. Thus, the molecular heterogeneity present within the tumours provide a fitness advantage under selective forces.
Non-specificity, narrow therapeutic index and significant toxicities of cytotoxic drugs have led to the development of targeted therapies which aim to interfere with defined molecular processes. Although impressive results have been obtained and tumour regression has been observed in many patients, constitutively active molecules, positive feedback loops and receptor crosstalk limits the efficacy of targeted drugs. For instance, being a major driver in PDAC initiation and a central mediator in a variety of signalling pathways, oncogenic KRas remains the most important therapeutic target among drug developers. However, all clinical attempts to directly interfere with KRas enzymatic activity have failed due to mutations locking KRas in a permanently active state [189]. Thus, rather than inhibiting enyzmatic activity, there have been attempts to inhibit KRas, mainly through inhibition of essential post-translational modification by blocking FTase activity [190]. However, a Phase II study showed that the FTase inhibitor, 15777, did not exhibit single agent activity in advanced PDAC patients [190]. Additionally, in a Phase III trial with advanced PDAC patients, combination therapy with tipifarnib (a FTase inhibitor) and gemcitabine showed an unfavourable benefit-toxicity profile compared with gemictabine monotherapy, with no improvement in survival [191]. It has been suggested that compensatory increased geranyltransferase activity preserving KRas function is among the possible explanations for the clinical failure of FTase inhibitors [192]. The conclusion must be that, at the present time, KRas should be considered an undruggable target [22].
The constitutively active form of KRas also hampers EGFR-targeted treatments as has been shown in non-small-cell lung and colon cancer models [193,194]. In addition, cross-talk between EGFR/MET, EGFR/IGFR, constitutively activating mutations in the EGFR gene and receptor heterodimerisation of EGFR and receptor tyrosine-protein erbB-2 (ErBB2) play important roles in gaining resistance to EGFR inhibitors, such as erlotinib [179,195]. In addition, loss of PTEN expression is correlated with increased activation of AKT, which is a downstream effector of EGFR and ultimately resistance to erlotinib [196].
Regarding the central importance of IGF-1R signalling in PDAC, it seemed that the IGFR cascade might be a potentially good therapeutic target. However, although IGFR inhibitors have been shown to suppress growth of PDAC cancer cells in vitro, use of IGF-1R blocking antibodies (e.g. ganitumab) have been largely disappointing in clinical trials [197]. The most potent candidates for failure of IGF-1R targeted therapy is crosstalk with IR [46]. Thus, activation of IR-by IGF bypasses the IGF-1R and its inhibition [46]. Several other factors may confer resistance to IGF-1R-targeted therapies, including constitutive activation of downstream effector molecules (e.g., mutant BRAF and mutant KRAS) and receptor crosstalk with other membrane receptors including MET and EGFR [43,44,46].
Disappointing results were also obtained when Metformin was to used inhibit mTOR signalling. Metformin is a specific inhibitor of mitochondrial respiratory chain complex I, promotes LKB1-dependent activation of AMPK by reducing ATP production [198]. Nevertheless, mutations and polymorphisms in the LKB1 gene reduce the activation of AMPK and refractory to metformin leading to mTOR hyperactivation [198]. Interestingly, while rapamycin, a selective inhibitor for mTOR, suppresses IRS 1, it causes augmentation of PI3K/AKT activation in PDAC cells [198]. On the other hand, rapamycin did not have any stimulatory effect on ERK activation in PDAC cells [197]. Conversely, active site inhibitors of mTOR (KU63794 and PP242) abrogate AKT activation, whereas they increase ERK activation in PDAC [198]. These results imply that therapeutic effectiveness of mTOR inhibitors are diminished by activation of other upstream pathways, therefore, suppression of feedback loops should be a major consideration in the use of mTOR inhibitors for PDAC therapy to counter balance anti-proliferative effects of mTOR.
Dynamic communications between heterogeneous cell populations, the stroma and environmental selective pressures drive therapeutic resistance. Exceptionally, abundant stromal content and poor blood supply of PDAC, acting as a pharmacological barrier, contribute to the failure of the systemic therapy. Where interstitial pressure has reduced, so affecting shrinkage of tumour blood vessels and resultant blood supply, gemcitabine activity has the potential to be enhanced through the induction of a re-expansion of tumour blood vessels using PEGylated human recombinant PH20 hyaluronidase, as shown in a xenografted PDAC model [199]. Remarkably, combination treatment with gemcitabine and PEGPH20 improved median survival and decreased metastatic burden [199]. Furthermore, targeting the Shh pathway using IPI-926 increased in intra-tumoural vascular density and intra-tumoral concentration of gemcitabine resulted in increased cancer cell destruction [200]. Accordingly, the constitutive hedgehog (Hh) pathway blockade using either small-molecule antagonists or the Shh ligand-blocking antibody 5E1 was also reported to inhibit distant metastases in a human pancreatic xenograft model [199,200]. However, clinical trials of HH pathway antagonists combined with cytotoxic chemotherapy have yielded disappointing results [201-203]. Lee et al. [204] found in three distinct genetically engineered mouse models that genetic and pharmacologic inhibition of the HH pathway activity actually accelerated PDAC progression. Furthermore, they reported that the acute modulation of pathway activity regulates the balance between epithelial and stromal elements, with inhibition causing suppression of desmoplasia and accelerated growth of epithelial elements and activation causing stromal hyperplasia and reduced growth of the neoplastic epithelium [204]. Given the strong influence of the stroma in the pathobiology of the tumour, it is not surprising that ECM/integrin signalling protects tumour cells from drug-induced apoptosis [205,206]. Accordingly, a meta-analysis revealed that genes associated with the integrin-mediated cell-adhesion and drug-resistance pathway are frequently overexpressed in PDAC [206]. In addition to the ECM-regulated chemoresistance, PSCs can directly impact the response of cancer cells to chemotherapy. For instance, PSC secretions have been shown to contribute to chemoresistance through enhancing inflammatory signals [206] and the CXCL12/CXCR4 axis [208].
Lastly, it is important to note that immunotherapeutic approaches, such as immune checkpoint inhibitors such as those characterised by antibodies targeting the T-lymphocyte regulators cytotoxic T-lymphocyte protein 4 (CTLA 4) and programmed cell death 1 (PD 1), have demonstrated efficacy in a variety of solid tumours, where they have already received US Food and Drug Administration approval [209]. However, since PDAC has generally been considered a non-immunogenic malignancy, immunotherapeutic approaches focusing on overcoming T-cell immunologic checkpoints have not yet found to be successful in the management pancreatic cancer [206,209].
Recently, attention has been focused on targeting PanCSCs to find better ways to combat PDAC, because increased ratios of PanCSCs relative to other cancer cells are associated with short survival, resistance to chemo- and radiotherapy, recurrence of the disease and enhanced metastatic potential. Most chemotherapeutic agents adversely affect differentiated cancer cells that make up bulk of a tumour, but they are often ineffective against PanCSCs. Pothula et al. reported that gemcitabine administration to mice immediately caused PDAC cell death reducing tumour volume at the start of treatment, however, did not had any inhibitory effect on the metastatic spread [210]. The lack of an anti-metastatic effect and the paradoxical increase in metastasis with gemcitabine treatment was explained by the fact that gemcitabine treatment selects out a subpopulation of PanCSCs with increased migratory potential owing to an increase in EMT [210]. Among the characteristics of CSCs is a high expression of drug efflux pumps such as multi-drug resistance gene protecting cells from chemotherapeutic reagents [73]. Furthermore, CSCs have a remarkable capacity to repair DNA damage caused by alkylating agents and radiation [211]. Also, CSCs primarily exist in the G0 phase of the cell cycle and are thus resistant to cell cycle specific drugs such as 5-FU [212]. Importantly, ALDH+ PanSCs are were found to be resistant to chemotherapy-induced cell death and are highly tumourigenic [213]. Regarding the role of ALDH1 in cellular detoxification, it is not surprising that PanCSCs cells expressing high levels of ALDH1 accrue a survival advantage under cytotoxic chemotherapy [213].
New treatment strategies that target genomic instability of PDAC are currently being investigated. Germline or somatic mutations in the BRCA1 and BRCA2, ATM and other genes involved in homologous recombination may confer increased sensitivity to DNA-damaging cytotoxic agents such as platinum analogues but also render these tumours uniquely susceptible to PARP inhibitors that disable base excision DNA repair, representing a potential synthetic lethal therapeutic strategy [214]. Veliparib which is a potent oral irreversible inhibitor of PARP 1/2, have been evaluated in clinical trials in patients with known BRCA mutations or a very strong personal or family history of pancreatic, or BRCA-related malignancy, an indicator of the potential presence of these mutations [214,215]. The phase IB trial demonstrated that the triple combination of veliparib, cisplatin (a platinum-based drug) and gemcitabine was reported to show high activity in BRCA-related PDAC [215]. In contrast, no significant activity was observed in non-BRCA-mutated patients [215] The drug was fairly well tolerated except two dose-limiting toxicities including neutropenia and thrombocytopenida, mostly with 80 mg daily continuous dosing [215]. Currently, a randomized phase II trial study has been investigating how well veliparib together with gemcitabine and cisplatin work, as compared with gemcitabine and cisplatin, in treating advanced PDAC patients who are known carriers of BRCA or PALB2 mutations (ClinicalTrials.gov identifiers: NCT01585805). In addition, a phase III, randomised, double blind, placebo controlled, maintenance olaparib (PARP 1 inhibitor) monotherapy in patients with BRCA mutated metastatic pancreatic cancer whose disease has not progressed on first line platinum based chemotherapy is also under investigation (ClinicalTrials.gov Identifier: CANC-3716).
Currently, a phase II trial is analysing whether the addition of erlotinib to gemcitabine adjuvant chemotherapy offers improved survival as compared with gemcitabine alone following resection of PDAC (ClinicalTrials.gov Identifier: RTOG 0848). Given the complexity of directly targeting EGFR signalling in PDAC, drug-development efforts have also focused on downstream components of EGFR. In this regard, targeting STAT3, a downstream effector of EGFR, caused a significant reduction in tumour burden and delayed tumour progression with increased response to gemcitabine associated with a decrease in proliferating cells [216]. Furthermore, the combination of inhibitor for SRC kinase, downstream mediator of EGFR, and 5-FU decrease 5-FU-induced activation of EGFR/AKT pathway and substantially decreased both in vivo tumour growth and distant metastasis [216].
A recent strategy to improve the treatment efficiency of gemcitabine have been developed by using the phosphoramidate ProTide approach [217] Slusarczyk et al. [217] developed Acelarin (NUC-1031) by addition of a phophoramidate ProTide moiety to gemcitabine which enables: a) passive entry into the cell; b) by-passing of the reliance on transporters; c) reduced reliance on kinases for phosphorylation, and, d) reduced susceptibility to deamination. Acelarin generates high intracellular levels of the active agents and has been well tolerated by the patients [217,218]. Results from the Phase I/II study showed that Acelarin achieved a remarkable disease control rate of 78% in patients with advanced, rapidly progressing solid tumours, relapsed/refractory on prior chemotherapy, including gemcitabine [218]. In particular, a pancreatic cancer patient with liver metastases showed partial response on Acelarin, with 30% reduction in tumour volume, 92% reduction in CEA level and 73% reduction in CA 19-9 [218]. Global phase III studies with Acelarin are currently being planned in ovarian, biliary and pancreatic cancer.
Ruxolitinib, a JAK1/JAK2 inhibitor approved for use in patients with myelofibrosis based on improvements in survival and symptoms, was shown to attenuated cachexia progression in a pancreatic cancer mouse model [219]. In a recently reported randomized phase II trial (the Ruxolitinib in Patients with Refractory Metastatic Pancreatic Cancer [RECAP] study), the addition of ruxolitinib to capecitabine was compared with capecitabine plus placebo in patients who had experienced progression on gemcitabine-based therapy [220]. Although no difference in survival was observed in the entire study population, preplanned analysis in the subgroup of patients with elevated levels of C-reactive protein (CRP), a well-characterized and sensitive marker of systemic inflammation, revealed a statistically significant survival benefit for the ruxolitinib-containing arm [220]. These promising results have led to two registrational phase III trials, termed JAK1 and JAK2, specifically limited to patients with metastatic pancreatic cancer who have high baseline CRP levels (ClinicalTrials.gov identifiers: NCT02117479 and NCT02119663) [220].
Considering the strong link between metabolic diseases and development of PDAC, there has been a promising trend towards incorporating use of metformin and cholesterol-lowering medications in the management of PDAC. Favourably, use of cholesterol-lowering medications such as simvastatin and atorvastatin, after PDAC diagnosis has been found to be associated with longer survival in patients with non-metastatic PDAC [221]. There is also some clinical evidence that metformin can reduce the incidence of PDAC and improve outcome of diabetic patients with PDAC [222,223]. The current proposed anti-tumour molecular action of metformin is mainly associated with the inhibition of insulin/IGF1 signalling, downregulation of mTOR signalling, activation of AMPK, disruption of crosstalk between insulin and GPCR systems [49,224,225]. However, a randomised phase II trial demonstrated that the addition of metformin to gemcitabine and erlotinib does not improve the outcomes of patients with locally advanced or metastatic pancreatic cancer [226]. Additionally, a phase II trial combination of metformin and pactlitaxel for patients with gemcitabine-refractory advanced did not find any benefit of adding metformin [227].
Given the fact that current chemotherapeutic agents appear to be largely ineffective at depleting the CSC pool, their combination with a CSC-targeted agent may promote tumour regression. Expectantly, combination of Notch signalling pathway inhibitor (PF-03084014) with gemcitabine was effective in inducing apoptosis, inhibition of tumour cell proliferation and angiogenesis, resulting in the attenuation of primary tumour growth as well as controlling metastatic dissemination, compared to gemcitabine treatment in a highly aggressive orthotopic model [228]. A triple combination treatment, comprising sonic HH inhibitor (cyclopamine/CUR199691), mTOR signalling inhibitor (rapamycin) and gemcitabine, was capable of eliminating PanCSCs in mice with established patient-derived pancreatic tumours [229]. Encouragingly, this combination was reasonably tolerated and translated into significantly prolonged long-term survival [229]. Accordingly, Sancho et al. [230] showed that PanCSCs are vulnerable to mitochondrial-targeted therapies (i.e., metformin) due to their strong reliance on oxidative metabolism. However, resistant clones eventually emerged during metformin treatment due to their intermediate glycolytic/respiratory phenotype. Sancho et al.[230] demonstrated that genetic/pharmacological targeting of Myc prevents/restores the response to metformin in resistant PanCSCs. Notably, verapamil, the calcium channel blocker, has been demonstrated to increase the cytotoxic effects of chemotherapeutic drugs and multi-drug resistance by targeting the transport function of ABC transporter B family member 1 (ABCB1) and ENT1 which are selectively overexpressed in PanCSCs [231].
A number of strategies are being investigated to target the stroma. Among them nab-paclitaxel appears promising because it combines cytotoxic therapy with targeted delivery. Nab-paclitaxel is a colloidal suspension of nanoparticles homogenised in human serum albumin bound to paclitaxel that is a microtubule-stabilizing agent inducing cell cycle arrest and ultimately cell death [232]. SPARC binds albumin and facilitate the delivery to the tumour of albumin-bound therapeutics [232]. Not surprisingly, PDAC with higher expressions of SPARC were associated with improved survival for patients who received the combination of nab-paclitaxel and gemcitabine in early clinical trials [233]. Nab-paclitaxel also decreases the content of PSCs, leading to reduced tumour stiffness [234]. Similarly, use of biphosphonates targeting PSCs caused a reduction in fibrosis, tumour volume/weight, peritoneal dissemination, angiogenesis and cell proliferation and increased apoptosis in an murine PDAC model [235]. These in vivo anti-tumour effects were enhanced when biphosphonates were combined with nab-paclitaxel [235]. Surprisingly, while PSCs-released HGF inhibition was effective in inhibiting local tumour growth, tumour angiogenesis and metastasis, anti-metastatic effect of HGF inhibition was lost when combined with gemcitabine [236]. It seems that the ability of gemcitabine to increase stemness as well as migratory potential of cells overcomes any anti-migratory influence of HGF inhibition [236]. Thus, targeted therapies will require careful modelling for optimal integration with existing treatment modalities.
Considering the high impact of inflammation on the initiation and development of PDAC, approaches to activate the specific immune-system components and overcome immune evasion seems to be promising. Appropriately, prevention B cell activity by ibrutinib, an inhibitor for Bruton’s tyrosine kinase and use of an agonist CD40 antibody reverse immune suppression and drive T cell-dependent anti-tumour responses suppressing tumour growth and improving responsiveness to standard-of-care chemotherapy [237-239]. Encouragingly, CD40 agonist antibody in combination with gemcitabine was reported to be well tolerated and associated with anti-tumour activity in patients with metastatic PDAC [240]. Similarly, inhibition of CXCR4 and blockage of CXCL12 receptor hold promise for a pharmacologic approach to increase the efficacy of immune checkpoint inhibitors such as anti–CTLA 4 and anti–PD 1 antibodies [241]. Currently, safety of continuous IV administration of plerixafor (a CXCR4 antagonist) patients with advanced pancreatic cancer is under investigation (ClinicalTrials.gov Identifier: CAM-PLEX, NCT02179970).
An alternative immune-based strategy under active clinical investigation in metastatic pancreatic cancer consists of a live-attenuated Listeria monocytogenes vaccine vector (CRS-207; Aduro Biosciences, BERKeley, CA) [242,243]. This bacteria has been genetically engineered to include mesothelin, a tumour associated antigen expressed in the majority of pancreatic cancers [241,242]. In the phase I trial, this agent was shown to induce mesothelin-specific T-cell responses, and three patients with pancreatic cancer who had undergone prior treatment with the cellular allogeneic pancreatic cancer vaccine (GVAX) in a separate study demonstrated markedly prolonged survival [242]. On these bases, a randomized phase II trial was recently conducted in patients with chemotherapy-refractory metastatic pancreatic cancer, demonstrating a statistically significant improvement in OS when sequential treatment with GVAX/CRS-207 /Cy (cyclophosphamide: inhibitor of Treg) was administered as compared with GVAX /Cy [243]. A larger randomized study, termed the Safety and Efficacy of Combination Listeria/GVAX Pancreas Vaccine in the Pancreatic Cancer Setting (ECLIPSE) trial, is now under way (ClinicalTrials-.gov identifier: NCT02004262).
Recently, McEwan et al.[244] have demonstrated the effectiveness of sonodynamic therapy (SDT) for the treatment of PDAC in a pre-clinical model. Basically, SDT involves the localised activation of an otherwise non-toxic sensitiser by a stimulus generating cytotoxic ROS [244]. Given the fact that PDAC is highly hypoxic, this negatively impacts the efficacy of the SDT approach. Thus, McEwan et al.[244] aimed to incorporate oxygen in the core of the microbubbles (MB) to enhance the amount of ROS generated in the tumour microenvironment, as oxygen is a substrate for ROS production in SDT. Encouragingly, an injection of ultrasound-responsive microbubbles (MB), filled with gaseous oxygen provided a significant SDT-mediated reduction in tumours. Later, McEwan et al. [244] has described the preparation of an oxygen-loaded microbubble (O2MB) platform for the highly targeted treatment of PDAC using both sonodynamic therapy (SDT) and anti-metabolite therapy (5-FU). The aim of this study was to increase efficacy of anti-metabolite therapy by sensitising microenvironment and also reduce chemotherapy-associated side effects [245]. Remarkably, combining sonodynamic and anti-metabolite therapy using O2MB conjugates provided enhanced cytotoxicity in PDAC cell lines cultured and decreased ectopic cells tumour volume [245]. This approach is a promising alternative to chemotherapy for the treatment of locally advanced PDAC and could be effective as a neo-adjuvant therapy.245
In recent years, focus has shifted towards exceptional responders, the rare patients who has a surprising, dramatic response to a drug. Exceptional response is defined by National Cancer Institute (NCI) as a complete response or a partial response that lasted at least 6 months to a systemic treatment that was not expected in more than 10% of patients [246]. The NCI has launched the Exceptional Responders Initiative in September 2014, which attempt to understand the molecular underpinnings of exceptional responses to treatment, primarily via chemotherapy, in cancer patients (ClinicalTrials.gov Identifier: NCT02243592, estimated primary completion date: January 2100). One specific report has demonstrated the feasibility of this approach [247]. Iyer et al.247 ran a whole-exome sequencing (WES) analysis of the bladder cancer tissues from exceptional responders who received the everolimus, an mTOR inhibitor. The biological mechanism behind this exceptional respond was correlated with loss-of-function mutations in TSC, a negative regulator of mTOR, in the tumour from the patients who showed good tumour response (tumour shrinkage) in that trial, whereas in the absence of TSC1 mutations, tumours did not shrink [247]. From this point of view, TSC1 was hypothesized to be a predictive response marker for everolimus treatment, leading to a prospective trial to test everolimus therapy for patient with TSC1-mutated tumours [246, 247]. Other exceptional responses were reported by Subbiah et al.[248] who demonstrated two patients with Ewing’s sarcoma remarkable responses to single-agent IGF1R therapy and then relapse. Once resistance to single-agent IGF1R inhibitor developed, patients were subjected to combined IGF1R plus rapamycin.248 Clinically, both patients responded to the combination therapy [248]. Upregulation of phosphorylated (p)-AKT and p-mTOR occurred in the resistance tissue, demonstrating the AKT/mTOR pathway to be the mechanism of acquired resistance to single-agent IGF1R therapy [248]. While one patient continued to respond, the second patient progressed [248]. Further analysis showed that the resistant tumour that emerged showed concurrent activation of the ERK pathway as a potential mechanism of resistance [248]. These findings emphasis the fact that cancer treatments require a moving, dynamic target planning which can be possible by rapid study of tumour tissue of patients virtually in real-time.
Fortunately, exceptional responses have also been reported in PDAC. Chue et al. [249] reported of a patient with metastatic PDAC who responded extremely well to (five years from diagnosis until report published) treatment with metronomic dosing of POLF (paclitaxel+oxaliplatin+leucovorin+5-FU) and a gemcitabine-based regimen, including oxaliplatin and leucovorin/5-FU with intemittent cetuximab (EGFR inhibitor). It is noted that the patient’s tumour showed no KRAS mutation indicating that wild-type KRAS responds to cetuximad and contributes to patient’s unusual long survival [248] Klimant et al. [250] presented a case of a patient with PDAC metastatic to the liver who experienced gemcitabine-based chemotherapy. The regimens included cycles of gemcitabine and oxaliplatin (GEMOX), followed by cycles gemcitabine, docetaxel and capecitabine and then cycles of gemcitabine and nab-paclitaxel, with an exceptional response 2 years from the initiation of chemotherapy for metastatic pancreatic cancer [250]. Although genetic testing were not performed in this patient, patient’s exceptional response to these treatments may lead to the conclusion that this patients’ tumour may have had a unique genetic and molecular makeup. Noteworthy, Rios Perez et al. [251] reported a case of a 50-year-old woman who presented with primary PDAC with two liver metastasis and high CA 19-9 level. The patient received six cycles of FOLFIRONIX over three months with removal of oxaliplatin the last month [251]. A dramatic reduction in CA 19-9 and complete regression of both liver metastases were observed [251]. With no evidence of distant disease, concurrent systemic and local therapy with capecitabine-based chemoradiation was performed with normal CA19-9 for eight months [251]. A mild increase in CA 19-9 promoted a restaging demonstrating an active primary tumour but no distant disease [251]. Then, a Whipple was performed rendering this patient free of detectable cancer [251]. A downstaging from a stage IV initially to stage IIB at the time of resection has generated 28 moths of survival to date of publication [251]. Ex-vivo chemosensitivity assay, in which the tumour tissue from the patient was examined for sensitivity to treatment regimen, demonstrated that this patient’s tumour sample was very sensitive to the FOLFIRINOX when compared to gemcitabine. This assay is under development as part of a clinical trial that will prospectively tests ability to identify the most effective treatment approach for individual patients [251].
Although promising, this approach has also several challenges. First, for each described mechanism for exceptional response, it is important to prove that the tumours in which they occur are truly dependent on the alterations and it is crucial to characterise the functional significance of such alteration. Second, it is often difficult to test dependency of the tumour to particular alteration, and find an agent or combination of agents specifically targeting particular alterations. Thus, the generation of patient-driven cell lines or patient-derived xenografts may eventually enable more definitive assessment of dependencies linked to specific genomic and phenotypic alterations in patient with the potential to be exceptional responders to anti-cancer agents. Meanwhile, exceptional responder cases may also prompt the identification of molecular mechanisms of therapeutic resistance. In addition, designing clinical trials with exceptional responders will allow more rapid discovery of pathways in cancer biology, and development of new agents or guide the use of existing cancer therapies. Importantly, this approach will help to predict major clinical responses to anti-cancer agents.
Currently, there is an observational study, underway on patients with metastatic pancreatic cancer to identify genomic differences between exceptional responders and non-responder to standard care chemotherapy (ClinicalTrials.gov Identifier: NCT02555735).
These examples highlight the importance of personalised medicine, which instruct molecular analysis of all tumours and help identify the most appropriate combination therapies.
As briefly reviewed earlier, current clinical management of PDAC relies largely on standard protocols. However, the use of same therapy approach for to all patients will result in variable and unpredictable responses, because of heterogeneity among tumours, and genotypic differences between patients influencing drug metabolism, drug transport, and an individual’s sensitivity to a drug and the patient’s epigenetic background. Hence, Personalised Medicine (PM) offers an attractive approach for PDAC-treatment and –care.
PM may be defined as “a form of medicine that uses information about a person’s genes, protein, and environment to prevent, diagnose, and treat disease” (National Cancer Institute 2011). PM implements “-omic” sciences (genomics, epigenomics, transcriptiomics, proteomics, metabolomics, etc.) to integrate various data sets with the aim of dissecting molecular signatures and functional pathways that help to classify tumour subtypes and determine their natural course, prognosis, and responsiveness to therapies. However, to ensure a truly individualised approach, genetic and epigenetic heterogeneity in time, microbiota, and a wide range of environmental factors including nutrition, stress and other factors modulating disease and response to treatment, must be considered. In this regard, personalised lifestyle medicine can be integrated throughout an individual’s life, whether for prevention or the management of treatment of PDAC.
The success of a personalised approach to PDAC management is dependent on the existence of reliable biomarkers Ideally, this should include early detection strategies with non-invasive screening programs using diagnostic biomarkers, collaborative decision-making involving patients and secondary care physicians and their support staff informed by prognostic biomarkers, delivery of individualised, multi-factorial treatment and monitoring regimens guided by predictive biomarkers, as well as ongoing monitoring of patient outcomes, using biomarkers and quality of life markers (Table 6, Figure 5).
Table 6. Potential application of personalised medicine in the management of PDAC.
Goal |
Approach |
Overall rationale/comments |
|
Diagnosis |
Minimal/non-invasive multiple biomarker approach with high specificity and sensitivity |
Multiple biomarker approach to increase effectiveness early detection and surveillance. |
Diagnostic BM panels |
Diagnostic sample |
Sensitivity (%) (HC vs PDAC) |
Specificity (%) (HC vs PDAC) |
3-protein panel (TNC/TFPI/CA19-9) [303] |
Plasma |
97 |
90 |
3-protein panel (CA 19-9, ICAM1, OPG) [304] |
Serum |
78 |
94 |
3-protein panel (CA 19-9, CEA, TIMP1) [304] |
Serum |
71 |
89 |
3-protein panel (LYVE1, REG1A, TFF1) [305] |
Urine |
77 |
90 |
miRNA panel (-21,-210, -155, -196a) [306] |
plasma |
64 |
89 |
5-CpG sites (IL10_P348, LCN2_P86, ZAP70_P220, AIM2_P62,TAL1_P817) [307] |
Circulating leukocytes |
65 |
90 |
25-cytokines +complement panel (C1 esterase inhibitor, C3, C5, CD40, Eotaxin, GM-CSF, IgM, IL-11, IL-12, IL-16, IL-1α, IL-1-ra, IL-2, IL-3, IL-4, IL-7, Integrin α-10, MCP-1, -3, Mucin-1, Properdin, TGF-α, TGF-β1, TNF-β, VEGF) [308] |
Serum |
73*
*Non-PDAC (HC, AIP, CP) vs PDAC |
75 *
*Non-PDAC (HC, AIP, CP) vs PDAC |
Autoantibodies panel (Anti-CTDSP1, -MAPK9 and -NR2E3 IgG) [309] |
Serum (higher levels of autoantibodies than controls) |
mtKRAS, mtTP53, mtSMAD4, aberrant DNA methylation 7-gene panel (FOXE1, NPTX2, CLDN5, CDKN2A,TFPI2, SPARC, ppENK) [20] |
Familial and sporadic PDAC tissues. (useful in screening for early non-invasive neoplasms). |
|
Therapy |
Potential personalised integrative therapeutic strategies |
|
Drugs and mechanism of action |
Predictive BM and clinical impacts for drugs |
Prognostic BM |
Natural compounds potentially targeting BM
|
Combinatorial treatment with natural compounds and chemotherapeutic drugs to promote therapeutic synergy and ultimately improve overall outcome |
- Gemcitabine: blocks DNA synthesis
- Gemcitabine+knockdown of STAT3: increases response to gemcitabine [216]
- Gemcitabine+HGF inhibition: decreases stemness, and migration [236]
|
- SNPs of CMPK1, CDA, dCK, RRM1, RRM2, ENT1, SLC29A1 are associated with response to gemcitabine [310-313]
- ↓ENT1, ↓CNT1, ↓CNT3, ↓dCK ↓TK2, ↑RRM1, ↑RRM2 expressions are associated with resistance to gemcitabine [174-177].
- ↓p53, ↓PTEN, ↑Bcl family proteins, ↑NF‑κB and ↑Hif-1α, ↑SRC, ↑EGFR, ↑STAT3, ↑PI3K/AKT, ↑Notch, ↑MAPK, ↑HGF pw are associated with resistance to gemcitabine [178-184,236]
- ↑miR-21, ↓miR-200, ↓let-7 are associated with resistance to gemcitabine [180,289]
|
- SNPs of CMPK1, CDA, dCK, RRM1, RRM2, ENT1, SLC29A1 are associated with grade 3/4 neutropenia and PFS [310-312]
- ↑miR-21, ↓miR-200 are associated with aggressiveness of PDAC cells [180,289]
|
- Curcumin and analogs, capsaicin, flavonoids, isothiocyanates, resveratrol.
|
- Curcumin or capsaicin, or flavonoids, isothiocyanates, or resveratrol treatments in combination with gemcitabine reverses gemcitabine resistance, suppress proliferation, invasion, angiogenesis and metastasis and increase apoptosis [180,289,314-317]
|
- 5-FU: inhibits TS, RNA and DNA synthesis
- PP2: SRC kinase inhibitor
- 5-FU+PP2: decreases the 5-FU-induced activation EGFR/AKT pw.
|
- H3K4me2, H3K9me2, H3K18ac modifications are associated with response to gemcitabine [258]
- ↑TS, ↑DPD, ↓CNT1, ↑MRPs and are associated with resistance to 5-FU [179,187,188,318]
- ↑EGFR/AKT/SRC pathway is associated with resistance to 5-FU [185].
|
- ↓H3K4me2, ↓H3K9me2, ↓H3K18ac are each associated with poor survival [258]
- ↑DPD, ↑TS, ↑p-Src are each associated with poor survival [185]
|
- Flavonoids, resveratrol, capsaicin isothiocyanates curcumin, folate, folinic acid, vits B6, B12 and methionine.
|
- Flavonoids+5-FU treatment inhibits proliferation, and induces of apoptosis [294]
- Curcumin or flavonoids or isothiocyanates act as histone deacetylase inhibitors which might work in synergy with 5-FU [258,315,316]
- Folinic acid enhances the effects of 5-FU by inhibiting TS resulting in pronounced and prolonged inhibition of DNA synthesis [320]
- Curcumin or capsaicin or flavonoids or isothiocyanates or resveratrol downregulating EGFR, AKT, SRC signallings might increase anti-tumour activity of 5-FU [315-316]
|
|
- Platinum-based agents (e.g., oxaliplatin, cisplatin) damage DNA.
- Veliparic, olaparib: PARP inhibitors
- Platinum agents + PARP inhibitors:
represents a synthetic lethal therapeutic strategy
|
- Deficiencey in “RADs, ATR, ATM, CHK1, CHK2, BRCA1, BRCA2, FANCG, FANCC, PALB2, ERCC1” are associated with improved response to platinum agents and PARP inhibitor sensitisation [60,215,321]
- ↑PER2 is associated with improved response to cisplatin [301]
|
- SNP of RAD54 is associate with reduced survival [322]
|
- Flavonoids, isothiocyanates
|
- Isothiocyanates act as a possible treatment mechanism to supplement PARP treatment [295]
- Flavonoids use in combination with cisplatin induce apoptosis [303]
|
|
- FOLFIRINOX : (5-FU, leucovorin, irinotecan, oxaliplatin)
- Irinotecan: Topoisomerase 1 inhibitor
- Leucovorin: Folinic acid
|
- FOLFIRINOX is more effective in patients with normal ERCC1 levels than in those with ERCC1 hyper-expression [321]
|
|
|
- Folinic acid increases anti-tumour activity of 5-FU [320]
|
|
- EGFR/MET, EGFR/IGFR, EGFR, erbB-2/EGFR heterodimerisation ↓PTEN, ↑AKT are associated with resistance to erlotinib [179,195,196]
- wtKRAS, ↑EGFR are associated with response to erlotinib [323]
|
- wtKRAS is associated with an improved OS in erlotinib-treated PDAC [323]
- mtKRAS is associated with reduced survival [23]
|
- Flavonoids, curcumin isothiocyanates, capsaicin, resveratrol.
|
- Curcumin, capsaicin, flavonoids, isothiocyanates, resveratrol downregulating EGFR, AKT, SRC signallings might increase anti-tumour activity of erlotinib [315,316]
|
- nab-paclitaxel: a microtubule-stabilizing
|
- SPARC enhances the delivery of nab-paclitaxel into tumour [324]
|
- ↑SPARC in the peritumoural stroma is related with worse prognosis [324]
- ↑SPARC is associated with improved survival for patients who received the combination of nab-paclitaxel and gemcitabine [324]
|
- Retinoic acid, and vit A, vit D3
|
- Use of retinoic acid, or vit A, or vit D3 might reduce activity of PSCs to synergise the effect of nab-pactitaxel and reduce tumour stiffness [234,315,316]
|
|
Personalised life style recommenda-tions |
Dietary interventions
|
Physical activity interventions
|
Mind-body interventions
|
Environmental interventions
|
Application of integrative oncology for achieving optimal outcome and maximal QOL
|
- Calorie restriction [280]
- Ketogenic diet [286]
- Limiting food access to the dark phase (8-9h access in the active phase) [302]
- A diet including high consumption of fresh fruits and fresh vegetables, Vit C, E, omega-3 fatty acids, and low consumption of red meat, saturated fats, processed foods, added sugar, soft drinks, and sweetened fruit soups or stewed fruit [272]
|
|
- Meditation, [269,270]
- Scheduled exercise [274]
- Yoga [274]
|
- Limiting alcohol consumption to 1 drink per day [272]
- Quit smoking,
- Avoiding tobacco smoke; infectious agents; radiation; industrial chemicals, pollution and medication.
|
- Time restricted feeding to improve glucose tolerance and nutrient homeostasis, and reduce insulin resistance, whole fat accumulation and inflammation.
- A healthy diet,
- Increased physical activity,
- maintain healthy weight,
- Avoiding external and internal toxicants
- Well-timed circadian/sleep cycles to rescue body clock and maintain metabolic health
- Relaxation strategies to reduce stress, improve mood, alter health behaviour, and maintain adherence to cancer treatment and a reduction in smoking.
|
( BM: biomarkers: Mt: mutation; wt: wild-type; HC : healthy controls; PDAC; pancreatic ductal adenocarcinoma; AIP: autoimmune pancreatitis; CP: chronic pancreatitis CA 19-9: cancer antigen 19-9; TNC: tenascin C; TFPI: tissue factor patway inhibitor; ICAM1: intercelular adhesionn molecule 1; OPN: osteopontin; CEA: carcinoembryonic antigen; TIMP-1: metalloproteinase inhibitor 1; LYVE1: lymphatic vessel endothelial hyalunoric acid receptor 1; REG1A: lithoastathine-1-alpha; TFF1: trefoil factor 1; C1 esterase inh.: C3: complement 3; C5: complement 5: CD40: cluster of differentiation 40; GM-CSF: granulocyte macrophage colony-stimulating factor; IgM: immunoglobulin M; IgG: immunoglobulin G; IL-11: interleukin-11; IL-12: interleukin-12; IL-16: interleukin-16; IL-1α: interleukin-1 apha; IL-1-ra: interleukin 1 receptor antagonist: IL-2: interleukin-2; IL-3: interleukin-3; IL-4: interleukin-4; IL-7: interleukin-7; Integrin α-10: integrin alpha-10; MCP-1: monocyte chemoattractant protein-1; MCP-3: monocyte chemoattractant protein-3; TGF-α: transforming growth factor alpha; TGF-β1: transforming growth factor beta-1: TNF-β: tumor necrosis factor alpha; VEGF: vascular endothelial growth factor; CTDSP1: carboxy-terminal domain: RNA polymerase II: polypeptide A small phosphatse 1; MAPK9: mitogen-activated protein kinase 9; NR2E3: nuclear receptor subfamily 2 group E member 3; SNP: single nucleotide polymorphism; CMPK1: cytidine/uridine monophosphate kinase 1; CDA: cytidine deaminase; dCK : deoxycytidine kinase; RPM1: ribonucleotide reductase 1; RPM2: ribonucleotide reductase 2; ↑: increase; ↓: decrease; ENT: equilibrative nucleoside transporter ; SLC29A: slute carrier family; STAT3: signal transducer and activator of transcription 3; HGF: hepatocyte growth factor; pw: pathway; MAPK: mitogen activated kinase: NF–kB: nuclear factor-kappa-B; PTEN: phosphatase and tension homolog; AKT2: v-AKT thymoma viral oncogene homolog 2; PI3K: phosphoinositide-3-kinase; Hif-1α: hypoxia-inducible factor 1-alpha; H: histone; me2: dimethylation; ac: acetylation; K: lysine; R: arginine; PFS: progression free survival; 5-FU: fluorouracil; thymidylate synthase (TS); EGFR; epithelial growth factor receptor; CNT1: nucleoside transporter; MRPs: multiple drug resistance-transporters; DPD: dihydropyrimidine dehydrogenase: Vit: vitamin; DNA: deoxyribonucleic acid; e.g.: example; PARP: poly(ADP-ribose) polymerase; ATR: ATR serine/threonine kinase; ATM: ATM serine/threonine kinase; CHK1: checkpoint kinase 1; CHK2: checkpoint kinase 2; BRCA1: breast cancer type 1 susceptibility protein; BRCA2: breast cancer type 2 susceptibility protein; FANCG: fanconi anemia complementation group G; FANCC: fanconi anemia complementation group C; PALB2: partner and localizer of BRCA1/2; ERCC1: excision repair cross-complementation group 1; PER2: period circadian clock 2; SPARC: secreted protein acidic and cysteine rich; PSC: pancreatic stellatte cell; p-SRC: phospho-SRC; OS: overall survival; QOL: quality of life; ↓: low expression; ↑: high expression
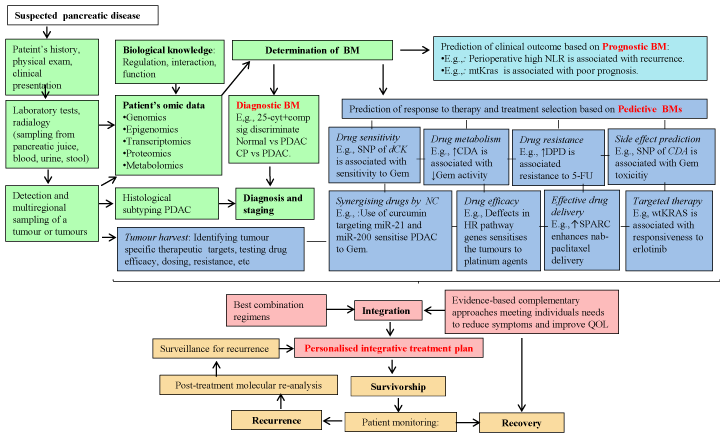
Figure 5. Schematic illustration of the potential applications of biomarkers for personalised integrative approaches in the management of PDAC. Combination of genetic and functional data into network modeling to predict phenotypic PDAC diversity to select an appropriate therapy for individuals and integrate the best evidenced-based and clinically validated complementary approaches meeting individuals needs to reduce symptoms and improve QOL.
(Abbreviations: BM, biomarker; vs, versus; wt, wild type; inh,inhibition; cyt+comp sig, cytokines, complemet signature; NLR, neutrophil-to-lymphocyte ratio; KRAS; Kirsten rat sarcoma viral oncogene homolog; Gem, gemcitabine; CDA, cytidine deaminase; CMPK1, cytidine monophosphate (UMP-CMP) kinase 1; DPD, dihydropyrimidine dehydrogenase; CDF, curcumin analogue; SNPs, single nucleotide polymorphisms; 5’-FU, fluorouracil; QOL, quality of life; dCK, deoxycytidine kinase; HR, homologous recombination; NC, natural compounds; ↓, low; ↑,high)
Timely detection of PDAC with accurate non-invasive biomarkers of early-stage disease can be achieved by screening high risk individuals. In particular, development of “multiple-biomarkers panels” to increase sensitivity of the screening test monitoring of individuals under risk are believed to have a great potential to increase diagnostic accuracy for early detection. A practical approach in selecting the screening population can be based on the epidemiological evidences, and clinical parameters including obesity, diabetes, chronic pancreatitis or hereditary PDAC syndromes. In addition to personal, family and genetic history, environmental factors should be considered for risk stratification and development of tailored screening and surveillance programs. For instance, considering the link between diabetes and PDAC, selected SNPs in metabolism-related genes such as IGF1, and ADIPOQ may provide an important screening tool to help identify individuals at increased risk of development of PDAC.
Clearly, the requirement of prior knowledge of the mechanism of resistance, early initiation of anti-cancer therapy while avoiding excessive toxicity has brought predictive biomarker to the centre of cancer therapy. However, due to less available tissues and heterogeneity between PDACs, determination of prognostic and predictive biomarker for planning treatments and surveillance strategies have been challenging. Indeed, the need to obtain metastatic tissues and tissue samples during different stages of the disease, as well as the changing landscape of tumour biology and growing resistant clones during therapy have hampered the development of effective treatments. Thus, use of circulating biomarkers appears to be preferable due to their ease of collection during the course of the disease and treatment and are relatively non-invasive in nature. Among them, auto-antibodies, circulating tumour cells (CTCs), tumour-released-protein, and -metabolites and tumour-derived extracellular vesicles (EVs), have gained attention during the past years. Importantly, the presence of auto-antibodies months to years before symptomatic disease develops, generates hope for developing useful diagnostic and prognostic tools. Notably, besides being a potential biomarker, CTCs hold promises for repeated study of tumour genetics, proteomics and molecular biology of the PDAC cells as well as pharmacodynamics throughout a patient’s clinical course. CTCs, having acquired the ability to enter the circulatory system, have been detected in peritoneal fluid and peripheral blood of 40%-100% PDAC patients [252,253]. Heterogeneous populations of CTCs allow phenotypic identification for treatment stratification [252]. Since, CTCs display tumour initiating capacity contributing to distant metastasis, detecting CTCs might increase the possibility of detecting metastasis at an early stage. This may improve prognosis following surgical resection by identifying patients who are appropriate candidates for early treatment with systemic therapy [252,253]. In addition, preoperative neutrophil-to-lymphocyte ratio (NLR) has been proposed a promising predictor of survival in patients with PDAC [254]. Remarkably, a novel, non-invasive three-protein biomarker panel that is able to detect patients with early-stage PDAC completely non-invasively, through analysis of urine samples have been established [255]. Sampling pancreatic juice or tumour during EUS could be of great clinical value providing biomarkers [256]. Strikingly, Stratford et al. [257] reported a six-gene signature that discriminates between high-risk (aggressive) and low-risk (less aggressive) tumours on the basis of patient survival. When researchers used this method with an independent set of tumour samples from patients with localised and resected PDAC, they found that patients with high-risk tumours had an average survival time of 15 months; 55% of them were alive a year after surgery, and the remaining, who had low-risk tumours, had an average survival time for 49 months; 91% of them were alive a year after resecrtion [257]. Regarding the risk of Whipple procedure (2%–6% of patients die during this operation and more than 50% have serious postoperative complications) the predictive ability of this signature might be used to help clinicians and patients make decisions about their treatment plan [257]. Histone profiling is also under investigation to draw a histone modification patterns for PDAC. A phase III clinical study by Manuyakorn et al. [258] demonstrated that cellular levels of histone modifications define previously unrecognized subsets of patients with PDAC with distinct epigenetic phenotypes and clinical outcomes and represent prognostic and predictive biomarkers that could inform clinical decisions, including the use of 5‘FU chemotherapy [258]. Furthermore, EVs that can be detected in peripheral blood, pleural effusions, and urine, offer the benefit of detecting and analysing molecular cargo of tumour origin such as miRNAs, mutated genomic fragments lipids, proteins as well as monitoring disease progression over-time [259]. For instance, Melo et al. reported increased expression of glypical in PDAC patients. Also, they found that glypican positive EVs were also detected prior to formation of PanIN lesions in mouse models of PDAC with increased proportionality over time. Additionally, detection of mutant Kras transcript in glypican-1 positive EVs highlight their potential utility identifying cancer specific genetic defects [260].
Recently, studies have focused on selecting and validating methods for stratifying tumours according to their molecular signature and allowing the identification of patients unlikely to derive clinical benefit from available chemotherapeutics. In this regard, Ghazaly, et al. [261] has reported that RNA-based detection method, namelyRNAscope technology, allows reliable detection of biomarkers, ENT1, CDA, dCK, thought to be associated with PDAC cells resistance to gemcitabine [308]. Currently, a clinical trial is under investigation aiming to discover possible biomarkers to predict additional benefit of acelarin over gemcitabine for subsequent validation (Acelarate, ISRCNT 16765355). In a retrospective study, Orlandi et al. [262]. compared gemcitabine versus FOLFRINOX in patients with metastatic PDAC in which ENT1 evaluation was available. Regardless of ENT1 expression, treatment with FOLFIRINOX revealed a statistically longer OS and PFS compared to gemcitabine [262]. However, when patients were stratified according to ENT1 expression, no differences in OS were found in ENT1 positive patients either treated with FOLFIRINOX or gemcitabine [262]. Strikingly, gemcitabine-treated ENT1 positive patients showed a statistically significant improvement both of OS and PFS in comparison to gemcitabine-treated ENT1 negative patients [262]. Thus, this study demonstrated the effectiveness of ENT1 in predicting gemcitabine activity and provides evidence for the use of ENT1 expression as a predictor for gemcitabine [262]. Since gemcitabine has a better safety profile than FOLFIRINOX, it may be more appropriate for ENT1 positive patients with advanced disease and poor performance. Recalling efficacy of platinum-based drugs in BRCA-related cancers, platinum based therapies in PDAC should be tailored to patients with tumours harbouring mutations in genes involved HR pathway.
A number of molecular targeted therapies, guided by specific biomarkers, have emerged in cancer chemotherapy, and show considerable promise. For instance, tumours overexpressing EGFR benefit from the EGFR-targeted agents [263]. Noteworthy, biomarkers where matched therapeutics already exist and can be “repurposed” or “rescued”, then cumulatively they provide a potentially significant opportunity to improve outcomes in a shorter timeframe than novel therapeutic discovery and development. Considering this, EGFR targeted therapies can be rescued in PDAC patients whose tumours bearing wild type KRAS and overexpressing EGFR. Evidently, in a randomized, open-label, prospective trial, adjuvant cemotherapy gemcitabine plus erlotinib was found to be more effective than gemcitabine alone for treating metastatic PDAC, especially those with EGFR mutations (ClinicalTrials.gov number, NCT01608841) [264].
Given the exceptional response of patients with TSC1 mutations using mTOR inhibitors, it may be valuable to evaluate repurposed mTOR inhibitors in a sub-population of PDAC patients with tumours harbouring genetic alterations in genes involved in LKB1, TSC1, and PTEN.
Clearly, determination of tumour sub-types according to biomarkers provides the opportunity to test the efficacy of targeted agents used in other types of cancers in PDAC patients where the tumour harbours the same molecular target. However, this strategy is still premature and will require extra care while applied. To address this, primary cell culture models appear to be testable to accurately define molecular portraits of the tumour and predict the therapeutic responses. Encouragingly, Golan et al. [265] developed a unique ascites-derived PDAC primary cell culture model to study signalling pathways in PDAC progression and to evaluate targeted therapies for the individual patients. Furthermore, Zhang et al.[266] have successfully has cultured PDAC organoids from patient biopsies for drug testing aiming to provides new personalised approaches to modelling and treating this malignancy.
Regrettably, cancer survivors are at a greater risk for developing other diseases and secondary cancers as a consequence of genetic predisposition, previous lifestyles, psychological/physical trauma, future lifestyle, and anti-tumour therapies. Furthermore, the decrease in functional status and increase in disability that result from cancer and its treatment greatly affect the QOL of cancer survivors. Research has shown that evidence-based complementary approaches used in conjugation with standard medical treatments may help to facilitate health during both active oncology treatment and survivorship [267,268]. Complementary therapies include massage therapy, acupuncture, mind-body therapies, musics, therapy, physical exercise and nutrition and nutritional supplements and other modalities [269,270] Integrative Oncology serves to optimally combine conventional therapies and the best complementary therapies to positive influence outcomes and improve quality of life, whether or not a person is near the end of his or her life [265-268]. With the evolution of personalised cancer care and growing evidence to support the efficacy of multi-factorial, integrative therapies, personalised treatment plans might be more likely than conventional treatments on their own to meet the patients’ needs, while also helping to improve survival and QOL of people affected by cancer. The application of biomarkers to clinical practice may not only facilitate the design of personalised therapies and better predict clinical outcomes, but also provide information to improve survival and QOL. Biomarkers that are modifiable by physical activity, diet, stress and environmental factors may be useful to help plan personalised lifestyles and to monitor responses to interventions. For instance, relevant biomarkers for the observed associations among physical activity, overweight or obesity, and cancer are sex steroid hormones, hyperinsulinemia and insulin resistance, metabolic hormones, increased inflammation, depressed immune function, and oxidative stress [271].
Since central adiposity and obesity are known risk factors for PDAC 272]. Exercise has a potential beneficial effect on tumour outcome by reducing insulin resistance and Insulin/IGF1 secretion. In addition, reduction in the adiposity will lead to a decrease in pro-inflammatory factors (e.g., TNF-alpha, IL-6, leptin) produced by adipocytes [273]. A recent randomized controlled trial revealed that patients with advanced PDAC who are strongly affected by fatigue, are likely to benefit from exercise interventions [274] However, due to multiple PDAC-related symptoms such as fatigue, depression, pain and malnutrition, exercise can be challenging for patients. Thus, adapted physical activity, which is a concept defined by the International Federation of Adapted Physical Activity (APA), has been suggested to patients with advanced PDAC in addition to usual care [274]. Implementation of an APA program includes individualising activities according to the patient physical fitness, exercise type preferences, psychological functions, and expectations), the cancer (stage, treatments, and tolerance), and the social environment [274].
Patients with PDAC often experience a severe pain, fatigue, anxiety and depression as well as fear and stress resulting in impaired QOL, reduced treatment adherence, and reduced survival [274,275]. Particularly, prolonged emotional distress leads to suppression or dysregulation of the immune system through downregulation of NK activity which is absent in PDAC [275]. In addition, stress can be co-factor for the initiation and progression of PDAC, since the catecholamine stress hormone (norepinephrine), which is also elevated by smoking, was shown to induce self-renewal and growth of PDAC cells and immortal ductal cells [276-278] Relaxation is associated with a reduction in the stress-induced psychological or physiological responses through modulation of cytokines and other mechanisms [277,278] Thus, relaxation strategies such as meditation, exercise, yoga, and other mind-body interventions can reduce stress, improve mood, alter health behaviour, and maintain adherence to cancer treatment and a reduction in smoking.
A considerable body of evidence suggests that excess oxidative stress causing cellular injury is a predominant mechanism that induces pancreatic inflammation leading to genotoxicity and cancer. Thus, minimising exposure to xenobiotics might be protective against PDAC. Briefly, the xenobiotic with greatest evidence of association with PDAC is tobacco smoke [10-12]. There is inconclusive evidence of associations between PDAC development and the following factors: infectious agents, radiation, certain industrial chemicals, airborne pollutants, some foodstuffs and some medications [10,11]. Generation of metabolites from processes such as inflammation, lipid peroxidation, oxidative stress, disease state, infection and microflora may be considered as an internal environmental factor and may be associated with PDAC risk [10,11].
Diets rich in fruit and vegetables are known to reduce risk of PDAC [272] Accordingly, in a large population-based case-control study revealed that greater intake of omega-3 fatty acids, vitamin C and E may reduce risk of PDAC [272]. Dairy foods have been proposed in various studies to both prevent and promote PDAC. However, in a recent pooled analysis of 14 cohort studies null associations were observed for dietary foods, calcium and total vitamin D intake during adulthood and PDAC risk [279]. High consumption of red meat, saturated fats, certain monounsaturated fatty acids and processed foods can cause or accelerate PDAC [10,272]. WCRF noted that red meat is a source of iron which can lead to the production of free radicals [272]. When cooked at high temperatures, red meat can also contain heterocyclic amines and polycyclic aromatic hydrocarbons [272]. In addition, if alcohol is consumed, it is recommended to be limited no more than 1 drink per day for women or per day for men [272]. Regarding involvement of impaired glucose metabolism in development of PDAC, modifying energy balance through controlling total calorie intake and increasing physical activity to manage body weight have been recommended for the purpose of either prevention or as of a treatment part of PDAC [272]. Especially, caloric restriction (CR), and ketegonic diet (KD), have become attractive tool for management of cancer. CR is a chronic regimen typically aiming to a reduce total caloric intake to a level 20-40% lower than that of a typical diet without limiting essential vitamins and nutrients. reduction in total energy intake but isonutrient vitamins, minerals, fatty acids, and amino acids relative to an ad libitum-fed control regimen [280]. CR, which prevents or reverse obesity, improves insulin sensitivity, and inhibits the development and progression of a variety of cancer including PDAC [280,281]. CR regimen in PDAC decreases circulating levels of leptin and IGF-1, reduces expression of Glut1, suppresses activation of Akt/mTOR, ERK, STAT3, NF–κB and activates AMPK and SIRT1 [281-283] KD, is described as a high-fat and low carbohydrate diet that elevates circulating levels of ketone bodies, serving as an alternative energy source [284] Shukla et al.[284] identified that ketone bodies revert metabolic adaptations in PDAC cells to induce growth arrest and apoptosis. Treatment with ketone bodies lead to a a reduction in glucose uptake, glycolytic flux, glutamine uptake, lactate secretion and ATP content in PDAC cells.284 Importantly, metabolic reprogramming of tumour cells by ketone bodies is responsible for diminishing cancer cell-induced cachexia in both cell line models and animal models of PDAC. 284 In addition, keeping in view the significant role of inflammation and metabolic alteration, a ketogenic diet also provides an efficient therapeutic strategy, because lowering lactate production by ketogenic diet has been shown to decrease MDSC frequency leading to improved anti-tumour immune response.285 Remarkably, a ketogenic diet causes minimal side effects as previously demonstrated that a 2-7 mM ketone body concentration can be achieved without giving rise to clinical acidosis.286 Recalling the fact that, at the time of diagnosis around 80% of patients with PDAC present cachexia, ketogenic diet might serve as an anti-cachectic agent as well as an anti-cancer agent.284-286
Considering the limitations of current conventional chemotherapeutics, including serious toxicities, development of chemoresistance and reduced QOL for cancer patients, the development of safe and efficacious alternatives with known and predictable mechanisms of action is much needed. Indeed, overcoming chemoresistance of PDAC cells should significantly extend patient survival. In this regard, combination therapy, with the goal of optimising cancer cell cytotoxicity while minimising systemic toxicity, appears the most rational primary clinical management strategy for PDAC. Unfortunately, most investigations have tended to be laboratory or animal-model rather than being clinically based. This is in part because patients with metastatic PDAC and poor performance status are typically excluded from clinical trials of new systemic treatments due to concerns that these patients may not tolerate the greater toxicity associated with combination chemotherapy regimens. Therefore, the use of combination therapy has prompted investigation into the use of natural compounds abundant in the diet as synergistic agents for exacerbating drug cytotoxicity in PDAC. In recent years, there has been increasing evidence that many natural compounds are effective in reversing, suppressing or preventing the initiation, promotion, or progression of cancer through targeting the genetic and/or epigenetic machinery and regulating signalling pathways. Thus, natural, plant-based compounds present in certain diets hold promise for chemo-preventive/-therapeutic interventions due to their potential ability to prevent and suppress cancer (Table 5).
Table 5. Natural products studied in relation to PDAC and their respective mechanism of action [315-317]
Compound (s) |
Natural source |
Mode of action |
Major targets |
Curcumin and analogs |
Curcuma longa (turmeric) |
↓inflammation, ↓proliferation, ↓invasion, ↓survival, ↓clonogenicity, ↑apoptosis |
↓NF-κB pw, ↓IL-8, ↓IL-8 pw, ↓COX-2, ↓STAT3, ↓Notch-1 pw, ↓PI3K/AKT/mTOR pw, ↓EGFR, ↓MAPK pw, ↑ATM/CHK1 pw, ↑TNFR pw, ↑caspases-8,3, ↑PTEN, ↓prostaglandin E2, ↓miR-21, ↑miR-200, ↓HDACs, ↓DNMT1 |
Capsaicin |
Chili peppers |
↓inflammation, ↓pain, ↓proliferation, ↑apoptosis, ↑ROS generation |
↓Bcl-2, ↓PI3K/AKT pw, ↓MAPK pw, ↓ Hedgehog pw, ↓NFκB pw, ↑MKK4 pw, ↑JNK pw, ↑caspase-3,9 |
Flavonoids
Epigallocatechin-3-gallate, kaempferol |
Fruits, vegetables, leaves, grains
Camellia sinensis
(green tea) |
↓inflammation, ↓proliferation, ↓EMT, ↓invasion, ↓migration, ↓metastasis, ↓self-renewal, ↓ clonogenicity,
↓angiogenesis, ↓anaerobic glycolysis, ↓glucose consumption, ↓lipogenesis, ↑apoptosis,
↑ROS generation |
↓NFκB pw, ↓JAK/STAT3 pw, ↓EGFR pw, ↓KRas/B-raf/MAPK pw, ↓PI3K/Akt/mTOR pw, ↓Notch pw, ↓ Hedgehog pw, c-↓Myc, ↓Nanog, ↓Oct-4, ↑c-JUN, ↓MMP-2,7,9,12 ↓VEGF, ↓IL-1, ↓IL-8, ↓IL-6, ↑caspase-8,3, ↑p21 pw, ↑JNK pw, ↓ LDHA, ↓AMPK pw, ↓HDACs, ↓DNMT pw, ↑p53 |
Isothiocyanates
|
Cruciferous vegetables |
↓proliferation,
↓self-renewal, ↓angiogenesis, ↑apoptosis,
↑genome stability |
↓Hedgehog pw, ↓NF-kappa-B pw, ↓AKT pw, ↓MAPK pw, ↓STAT3 pw, ↓HDAC pw, ↓MMP-2, ↓VEGF, ↓VEGFR pw, ↓Hif-1α, ↓STAT3 pw, ↓IL-6, ↑PARP, ↓HDAC1 |
Resveratrol |
Red grapes, peanuts, berries, dark chocolate, Japanese knotweed |
↓proliferation, ↓survival, ↑apoptosis, ↓EMT, ↓pluripotency,
↓migration, ↓invasion,
↓growth+↑apoptosis in acidic environment
|
↓ Hedgehog pw, MMP-2,9, ↓PI3K/Akt/NFkB pw, ↑p21, ↓Src pw, ↓STAT-3, ↑caspase-3/7, ↓Bcl-2, ↓Zeb-1, ↓Slug, ↓Snail, ↓Nanog, ↓c-Myc, ↓Sox-2, ↓Oct-4, ↓ABCG2, |
Folate, Vitamin B6, B12 and methionine
|
Whole grains, green leafy vegetables, oranges, and legumes |
↑DNA integrity, ↑DNA repair, ↑DNA methylation |
↑DNMTs,
↑S-adenosylmethionine |
Retinoic acid, and Vitamin A
|
Beef liver (rich), fruits (e.g., apricot, mango, orange, watermelon, blackberry, peach), vegetables (e.g., carrot, pumpkin, kale, spinach, sweet potato), cereals |
↓proliferation, ↓migration, ↓EMT, ↓fibrosis, ↑apoptosis
|
↓PSCs activity, ↓ECM, ↓IL-6, ↓Wnt pw |
Vitamin D3
|
Cod liver oil (rich), oily fish, dairy |
↓proliferation, ↓fibrosis, ↑apoptosis |
↓PSCs activity, ↓ECM |
IL: interleukin; NF–kB: nuclear factor-kappa-B; COX-2: cyclooxygenase 2; AKT2: v-AKT thymoma viral oncogene homolog 2; PI3K: phosphoinositide-3-kinase; STAT3: signal transducer and activator of transcription 3; MAPK, mitogen-activated protein kinase; PTEN: phosphatase and tension homolog; Hif-1α: hypoxia-inducible factor 1-alpha; EGFR: epithelial growth factor receptor; HDACs: histone deacetylases, DNMT: DNA methyltransferase; pw: pathway; JAK: janus kinase ATM serine/threonine kinase; CHK1: checkpoint kinase 1; MKK4: mitogen-activated protein kinase kinase 4; VEGF: vascular endothelial growth factor; TNF: tumor necrosis factor; MMP: matrix metalloproteinases, AMPK: AMP-activated protein kinase; PARP: poly(ADP-ribose) polymerase; SOX-2: SRY-Box 2; ECM: Extracellular matrix; EMT: epithelial mesenchymal transition; ABCG2: ATP binding cassette subfamily G member 2; PSC: pancreatic stellatte cell; ↓: low expression; ↑: high expression; e.g: example, ROS: reactive oxygen species.
For about a decade, gemcitabine monotherapy has remained the gold standard of treatment for patients with poor performance status. Thus, there has been interest in the use of natural products with potential for enhancement of the therapeutic potential of gemcitabine. In vitro studies demonstrated that epigallocatechin-3-gallate (EGCG) enhanced gemcitabine-induced apoptosis by activation caspase-3 and PARP and inhibiting JAK/STAT3 signalling [287] Ali et al.[180] showed that difluorinated curcumin (CDF), a curcumin analogue, reversed gemcitabine-resistance by increasing expression of miR-200 and decreased expression of miR-21 leading to reactivation of PTEN. They further demonstrated that CDF could sensitise PDAC cells to gemcitabine by inactivation of NF-κB and COX-2 [180]. Favourably, a phase I/II study showed that combination therapy using 8g oral curcumin daily with gemcitabine-based chemotherapy was safe and feasible in patients with pancreatic cancer, however further investigation into its efficacy is needed [288] Up-regulation of miR-200 and let-7 by isoflavone leads to the reversal of EMT in gemcitabine-resistant PDAC cells, which likely to be important for designing gemcitabine-based novel therapies for PDAC [289]. Importantly, in an orthotropic model of human PDAC, Harikumar et al. [290] found that resveratrol significantly suppressed the growth of the tumour and this effect was further enhanced by gemcitabine. The authors also demonstrated that resveratrol can potentiate the effect of gemcitabine through suppression of markers of proliferation, invasion, angiogenesis and metastasis [290]. A report by Arshad et al. [291] demonstrated that 100g intravenous marine-derived ω-fatty acids (ω-FA) in combination with gemcitabine shows evidence of improved activity and benefit to QOL in patients with advanced pancreatic cancer. The data from this study of gemcitabine and ω-FA infusion suggest that 47.2% of patients experienced a 10% or greater improvement in global health which is recognised as clinically significant and encouraging [291]. ESPAC-3 trial, phase III, randomized controlled trial conducted in 159 pancreatic cancer to determine whether 5-FU or gemcitabine superior in terms of OS as adjuvant treatment following resection of pancreatic cancer (clinicaltrials.gov Identifier: NCT00058201) [292]. Patients received folinic acid (20mg/m2) in combination with 5-FU or gemcitabine [292]. There were no significant differences in either PFS or global QOL scores between the treatment groups [292]. Compared with the use of fluorouracil plus folinic acid, gemcitabine did not result in improved OS in patients with completely resected pancreatic cancer [292].
Very recently, Cheng et al. [293]showed anti-proliferation effect of meisoindigo, chemically related to the natural product indirubin, on gemcitabine-resistant PDACs. In particular, cells bearing stem cell phenotype were found to be more vulnerable to meisoindigo, which reduces expression of CSC-associated genes, decreases cellular mobility and sphere formation and, lowers glucose uptake, while increases ROS level [293].
Kaempferol (ginkgo flavonoids) inhibits PDAC cell proliferation and induce apoptosis, and may sensitise cells to 5-FU, as their combinatorial administration has showed an additive effect on the inhibition of proliferation [294]. Notably, kaempferol was found to have significantly less cytotoxicity than 5-FU in normal human pancreatic ductal epithelial cells [294]. In addition, apoptotic cell population was increased when treated with kaempferol in a concentration-dependent manner [294].
Recent work implicates isothiocyanates, a key group of active ingredients in cruciferous vegetables, as a possible treatment mechanism to supplement PARP treatment [295]. In vitro studies by Banerjee et al. [296] demonstrated that that pre-treatment of cells with soy-derived genistein followed by cisplatin resulted in significant loss of cell viability and potentiated apoptosis irrespective of the metastatic ability of cells. In addition, the authors showed that genistein in combination with cisplatin was more an effective antitumor agent compared with cisplatin alone in their orthotopic tumour model [296]. Genistein sensitised pancreatic cancer cells toward cisplatin-induced apoptosis through suppression of Bcl-xL, Bcl-2, and AKT kinase and activation of NF-κB [294,296].
Activation of VDR is a critical determinant of cytotoxic drug sensitivity because of its importance to repairing stalled replication forks [297]. The mechanism of sensitization is via recruitment of RAD51, key protein in homologous recombination [297]. While VDR knockdown enhanced gemcitabine killing, increasing level of VDR expression and activation in PDAC cells increased their IC50 to gemcitabine [297]. The effects of VDR on gemcitabine sensitivity is ligand and dimerisation dependent as VDR mutants lacking these activities failed to the gemictabine sensitivity [297]. Inhibition of VDR in PDAC provides a way to enhance the efficacy of genotoxic agents such as PARP inhibitor and gemcitabine. In contrast, Persons et al [298] showed that alkylating derivate of vitamin D (1,25(OH)2D3), anti-proliferative property of Vitamin D is strongly enhanced by co-administration of AICAR, an activator of AMPK pathway, in pancreatic cancer cells. Hence, using specific natural or synthetic compounds in the management of cancer require a depth of understanding of the cross-talk between cancer signalling pathways and networks to retain or enhance chemopreventative activity or enhance effectiveness of chemotherapeutic reagents while reducing known toxic effects.
Chronotherapeutic approaches which rely on adequate circadian timing of cancer treatment schedules, provide encouraging results both in experimental and clinical studies by enhancing tolerability and efficacy of anti-cancer drugs. Application of chronobiological concepts to the treatment of PDAC appear to be effective as chrono-modulated infusional 5-FU chemoradiation has relatively less severe and less frequent acute toxicity than that reported with flat infusional or bolus 5-FU based chemoradiation used for adjuvant post-operative therapy for PDAC [299]. However, the optimal timing varies according to sex, genetic background, lifestyle, stage of cancer and presence of other diseases. To this extent, biomarkers related to the rhythmic features of drug metabolism, cellular detoxification, cell cycle and circadian rhythm may help to optimise timing drug administration. Respectively, the diagnostic and prognostic potentials of circadian genes in PDAC were reported, and their clinical utility is awaited to be validated in larger populations (Table 6) [300]. A supportive report by Oda et al [301]. suggested circadian gene PER2 as a predictive biomarker because the overexpression of circadian gene PER2. PER2 gene codes period circadian protein homolog 2, which acts as a tumour suppressor gene, and has synergistic effect with cisplatin (Table 6). Disruption of normal circadian cycle, causing dysregulation of circadian genes, has been associated with a variety of disease such as obesity, diabetes, inflammation, sleep disorders cancer including PDAC [300,301]. Thus, reprogramming circadian clock may provide a great opportunity to develop novel strategies aimed at treating or preventing circadian clock dysfunction along the course of cancer processes. Strikingly, in a mammalian PDAC model, meal timing was shown to induce rhythmic expression of critical genes and inhibit growth of tumour in spite of non-functional molecular clocks [302]. It seems that limiting food access to the dark phase, well-timed sleep cycles, scheduled exercise, a healthy diet (e.g., high in fruits, vegetables, whole grains) might effectively rescue the clock and help correct metabolic dysfunction. Such novel circadian-based supportive care deserves clinical testing.
Altogether, PDAC continues to pose a major therapeutic challenge as life expectancy rarely exceeds 12 months even in those patients who have a good performance status and are suitable for more intensified treatment regimens. This failure is largely due to pathobiological characteristics of PDAC and it is reasonable to speculate that a better understanding of PDAC biology may lead to development of effective management plans. Importantly, a better understanding of tumour biology together with the genetic predisposition to PDAC, gene-gene interactions, gene-environment interactions, and epigenetic phenomena will lead to identification of new targets and relevant pathways, serving as diagnostic, predictive and prognostic biomarkers. Thanks to novel technologies and “-omics”-based characterisation efforts, the molecular classification PDACs is evolving rapidly offering not only identification of multiple new targets, but also identification the subset of patients who might respond to particular combinations of therapies. Currently, surgery offers the best survival chance, thus, discovering accurate biomarkers for early diagnosis and identification of the patient subsets who are most likely benefit from surgery and neoadjuvant therapy might increase the number of resectable patients. Since, PDAC is a systemic disease already at the time of diagnosis management of PDAC requires a moving and dynamic planning which can be started with multiregional sampling of a patient’s tumours and combination of agents, with each agent targeted to the features of different sub-clones and microenvironmental compartment followed by monitoring the disease virtually in real time with circulating biomarkers and imaging modalities. Considering the limitations of current conventional chemotherapeutics, including serious toxicities and reduced QOL for cancer patients, the development of safe and efficacious supplementary or alternative interventions such as natural products with known/predictable mechanism of action seems to promote therapeutic efficacy while improving QOL. Further preclinical research followed by carefully designed clinical trials is highly necessary to accelerate the development of novel strategies offering best treatment to each patient. Personalised Medicine approach with lifestyle recommendations based on biomarkers may provide a novel means of assessing a PDAC patient’s health by empowering them with information they need to regain control of their life. It is hoped that incorporation of some of these approaches will go some way to improving QOL and survival for individuals with this devastating disease.
This article is dedicated to the memory of Jon Lord. We thank the Sunflower Jam charity for the award of the Jon Lord Fellowship to PK. We would also like to extend thanks to Meleni Aldridge (ANH International) for her encouragement and support throughout the duration of the project.
- GLOBOCAN 2012 (2014) Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 Lyon, France: International Agency for Research on Cancer.
- Klöppel G (2000) World Health Organization Classification of Tumours, Pathology and Genetics of Tumours of the Digestive System: Ductal adenocarcinoma of the pancreas, International Agency for Research on Cancer Press.
- Cancer Research UK (2014) Pancreatic cancer mortality statistics.
- Siegel RL, Miller KD, Jemal A (2016) Cancer statistics 2016. CA Cancer J Clin 66: 7-30.[Crossref]
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, et al. (2014) Projecting cancer incidence and deaths to 2030. The unexpected burden of tyroid, liver, and pancreas cancers in the United States. Cancer Res 74: 2913-2921. [Crossref]
- Spadi R, Brusa F, Ponzetti A, Chiappino I, Birocco N, et al. (2016) Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians. World J Clin Oncol 7: 27-43. [Crossref]
- Hootman SR, de Ondarza J (1993) Overview of pancreatic duct physiology and pathophysiology. Digestion 54: 323-330. [Crossref]
- Otsuki M, Tashiro M (2007) Chronic pancreatitis and pancreatic cancer, lifestyle-Related diseases. Intern Med 46: 109-113. [Crossref]
- Howlader N (2015) SEER Cancer Statistics Review, 1975-2012, National Cancer Institute.
- Maisonneuve P, Lowenfels AB (2015) Risk factors for pancreatic cancer: a summary review of meta-analytical studies. Int J Epidemiol 44: 186-198. [Crossref]
- Becker AE, Hernandez YG, Frucht H, Lucas AL (2014) Pancreatic ductal adenocarcinoma: Risk factors, screening, and early detection. World J Gastroenterol 20: 11182-11198. [Crossref]
- Bartsch DK, Gress TM, Langer P (2012) Familial pancreatic cancer-current knowledge. Nat Rev Gastroenterol Hepatol 9: 445-453. [Crossref]
- Aichler M, Seiler C, Tost M, Siveke J, Mazur PK, et al. (2012) Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: a comparative study in transgenic mice and human tissues. J Pathol 226, 723–734. [Crossref]
- Hruban RH, Klimstra DS (2014) Adenocarcinoma of the pancreas. Semin Diagn Pathol 31: 443-451. [Crossref]
- Erkan M, Michalski CW, Rieder S, Reiser-Erkan C, Abiatari I, et al. (2008) The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol 6: 1155–1161. [Crossref]
- Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, et al. (2012) The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol 9: 454-67. [Crossref]
- Ceyhan GO, Bergmann F, Kadihasanoglu M, Altintas B, Demir IE, et al. (2009) Pancreatic neuropathy and neuropathic pain-a comprehensive pathomorphological study of 546 cases. Gastroenterology 139: 177-186. [Crossref]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, et al. (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 32: 1801–1806. [Crossref]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, et al. (2015) Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518: 495-501. [Crossref]
- Brune K, Hong SM, Li A, Yachida S, Abe T, et al. (2008) Genetic and epigenetic alterations of familial pancreatic cancers. Cancer Epidemiol Biomarkers Prev 17: 3536–3542. [Crossref]
- Kanda M, Matthaei H, Wu J, Hong SM, Yu J, et al. (2012) Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 142: 730–733. [Crossref]
- Eser S, Schnieke A, Schneider G, Saur D (2014) Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 111: 817-822. [Crossref]
- Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, et al. (2012) Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 122: 639-653. [Crossref]
- Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, et al. (2012) Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One 7: e49707. [Crossref]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, et al. (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7: 469–483. [Crossref]
- Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, et al. (2012) A central role for RAF—>MEK—>ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov 2: 685–693. [Crossref]
- Grover S, Jajoo K (2016) Screening for Pancreatic Cancer in High-risk Populations. Gastroenterol Clin North Am 45: 117-127. [Crossref]
- Reznik R, Hendifar AE, Tuli R (2014) Genetic determinants and potential therapeutic targets for pancreatic adenocarcinoma. Front Physiol 5: 87. [Crossref]
- Morris JP, Wang SC, Hebrok M (2010) KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer 10: 683–695. [Crossref]
- Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D, et al. (2013) A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 145: 1449-1458. [Crossref]
- White, E (2013) Exploiting the bad eating habits of Ras-driven cancers. Genes Dev 27: 2065-2071. [Crossref]
- Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M (2013) Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology 144,1199-1209. [Crossref]
- Karna E, Surazynski A, Orłowski K, Łaszkiewicz J, Puchalski Z, et. al. (2002) Serum and tissue level of insulin-like growth factor-I (IGF-I) and IGF-I binding proteins as an index of pancreatitis and pancreatic cancer. Int J Exp Pathol 83: 239–245. [Crossref]
- Appleman VA, Ahronian LG, Cai J, Klimstra DS, Lewis BC (2012) KRAS(G12D)- and BRAF(V600E)-induced transformation of murine pancreatic epithelial cells requires MEK/ERK-stimulated IGF1R signaling. Mol Cancer Res 10: 1228–1239. [Crossref]
- Tanno S, Tanno S, Mitsuuchi Y, Altomare DA, Xiao GH, et al. (2001) AKT activation up-regulates insulin-like growth factor I receptor expression and promotes invasiveness of human pancreatic cancer cells. Cancer Res 61 589–593. [Crossref]
- Ruggeri BA, Huang L, Wood M, Cheng JQ, Testa JR (1998) Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog 2:81–86. [Crossref]
- Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, et al. (2005) The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF- kappa B and c-Myc in pancreatic cancer cells. Oncogene 23: 8571–8580. [Crossref]
- Morran DC, Wu J, Jamieson NB, Mrowinska A, Kalna G, et al. (2014) Targeting mTOR dependency in pancreatic cancer. Gut 63: 1481-1489. [Crossref]
- Feng Z (2010) p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb Perspect Biol 2: a001057. [Crossref]
- Rozengurt E, Soares HP, Sinnet-Smith J (2014) Suppression of feedback loops mediated by PI3K/mTOR induces multiple over-activation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther 13: 2477-2488. [Crossref]
- Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, et al. (2002) Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem 277: 39684–39695. [Crossref]
- Rozengurt E, Sinnett-Smith J, Kisfalvi K (2010) Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug amm in pancreaticcancer. Clin Cancer Res 16: 2505–2511. [Crossref]
- Ueda S, Hatsuse K, Tsuda H, Ogata S, Kawarabayashi N, et al. (2006) Potential crosstalk between insulin-like growth factor receptor type 1 and epidermal growth factor receptor in progression and metastasis of pancreatic cancer. Mod Pathol 19: 788-796. [Crossref]
- Ucar DA, Magis AT, He DH, Lawrence NJ, Sebti SM, et al. (2013) Inhibiting the interaction of cMET and IGF-1R with FAK effectively reduces growth ofpancreatic cancer cells in vitro and in vivo. Anticancer Agents Med Chem 13: 595-602. [Crossref]
- Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R (2009) Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev 30: 586–623. [Crossref]
- Ulanet DB, Ludwig DL, Kahn CR, Hanahan D. (2010) Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. PNAS 107: 10791–10798. [Crossref]
- Morton JP, Jamieson NB, Karim SA, Athineos D, Ridgway RA, et al. (2010) LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology 139: 586-97, 597.e1-6. [Crossref]
- Cheng X, Kim JY, Ghafoory S, Duvaci T, Rafiee R, et al. (2014) Methylisoindigo preferentially kills cancer stem cells by interfering cell metabolism via inhibition of LKB1 and activation of AMPK in PDACs. Mol Oncol [Crossref]
- Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E (2009) Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res 69: 6539-6545. [Crossref]
- Oliveira-Cunha M, Newman WG, Siriwardena AK (2011) Epidermal Growth Factor Receptor in Pancreatic Cancer, Cancers 3:1513-1526. [Crossref]
- Sánchez-González P, Jellali K, Villalobo A (2010) Calmodulin-mediated regulation of the epidermal growth factor receptor. FEBS J 277: 327-342. [Crossref]
- Tobita K, Kijima H, Dowaki S, Kashiwagi H, Ohtani Y, et al. (2003) Epidermal growth factor receptor expression in human pancreatic cancer: Significance for liver metastasis. Int J Mol Med 1: 305–309. [Crossref]
- Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, et al. (1998) Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res 58: 5329–5332. [Crossref]
- Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, et al. (1997) Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 57: 3126–3130. [Crossref]
- Kim WY, Sharpless NE (2006) The regulation of INK4/ARF in cancer and aging. Cell 127: 265-275. [Crossref]
- Ruggeri BA, Huang L, Berger D, Chang H, Klein-Szanto AJ, et al. (1997) Molecular pathology of primary and metastatic ductal pancreatic lesions: analyses of mutations and expression of the p53, mdm-2, and p21/WAF-1 genes in sporadic and familial lesions. Cancer 79: 700-716. [Crossref]
- Carnevale J, Ashworth A2 (2015) Assessing the Significance of BRCA1 and BRCA2 Mutations in Pancreatic Cancer. J Clin Oncol 33: 3080-3081. [Crossref]
- Powell SN, Kachnic LA (2003) Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 22: 5784-5791. [Crossref]
- Sy SM, Huen MS, Chen J (2009) PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA 106: 7155–7160. [Crossref]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, et al. (2006) Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 66: 8109-8115. [Crossref]
- Dong X, Li Y, Hess KR, Abbruzzese JL, Li D (2011) DNA mismatch repair gene polymorphisms affect survival in pancreatic cancer. Oncologist 16: 61-70. [Crossref]
- Swietach P, Hulikova A, Vaughan-Jones RD, Harris AL. (2000) New insights into the physiological role of carbonic anhydrase IX in tumour pH regulation. Oncogene 29: 6509-6521. [Crossref]
- Dovmark TH, Hulikova A, Vaughan-Jones RD, Swietach P (2014) Carbonic anhydrase activity regulates intracellular pH in 3D pancreatic cancer growths. Proc Physiol Soc 3: PCA114 (abstract).
- Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, et al. (2013) Acidity generated by the tumor microenvironment drives local invasion. Cancer Res 73: 524-1535. [Crossref]
- Kong SC, Giannuzzo A, Novak I, Pedersen SF (2014) Acid-base transport in pancreatic cancer: molecular mechanisms and clinical potential. Biochem Cell Biol 92: 449-459. [Crossref]
- Cardone RA, Greco MR, Zeeberg K1, Zaccagnino A2, Saccomano M, et al. (2015) A novel NHE1-centered signaling cassette drives epidermal growth factor receptor-dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia 17: 155-166. [Crossref]
- Yee NS, Chan AS, Yee JD, Yee RK (2012) TRPM7 and TRPM8 Ion Channels in Pancreatic Adenocarcinoma: Potential Roles as Cancer Biomarkers and Targets. Scientifica (Cairo) 2012: 415158. [Crossref]
- Dong H, Shim KN, Li JM, Estrema C, Ornelas TA, et al. (2010) Molecular mechanisms underlying Ca2+-mediated motility of human pancreatic duct cells. Am J Physiol Cell Physiol 299: 1493-1503. [Crossref]
- Ahmed N, Corey M, Forstner G, Zielenski J, Tsui LC, et al. (2003) Molecular consequences of cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in the exocrine pancreas. Gut 52: 1159–1164. [Crossref]
- Neureiter D, Jäger T, Ocker M, Kiesslich T (2014) Epigenetics and pancreatic cancer: pathophysiology and novel treatment aspects. World J Gastroenterol 20: 7830-7848. [Crossref]
- Visani M, Acquaviva G, Fiorino S, Bacchi Reggiani ML, Masetti M, et al. (2015) Contribution of microRNA analysis to characterisation of pancreatic lesions: a review. J Clin Pathol 68: 859-869. [Crossref]
- Fukushige S, Horii A (2014) Road to early detection of pancreatic cancer: Attempts to utilize epigenetic biomarkers. Cancer Lett 342: 231-237. [Crossref]
- Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414: 105-111. [Crossref]
- Li C, Lee CJ, Simeone DM (2009) Identification of human pancreatic cancer stem cells. Methods Mol Biol 568: 161-173. [Crossref]
- Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, et al. (2012) Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol Med 4: 218-33. [Crossref]
- Jimeno A, Feldmann G, Suárez-Gauthier A, Rasheed Z, Solomon A, et al. (2009) A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther 8: 310–314. [Crossref]
- Li C, Wu JJ, Hynes M, Dosch J, Sarkar B, et al. (2011) c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 141: 2218-2227. [Crossref]
- Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, et al. (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 313–323. [Crossref]
- Teicher BA, Fricker SP (2010) CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res 16: 2927-2931. [Crossref]
- Salnikov AV, Liu L, Platen M, Gladkich J, Salnikova O, et al. (2012) Hypoxia induces EMT in low and highly aggressive pancreatic tumor cells but only cells with cancer stem cell characteristics acquire pronounced migratory potential. PLoS One 7: e46391. [Crossref]
- Rausch V, Liu L, Apel A, Rettig T, Gladkich J, et al. (2012) Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol 227:325-335 (2012). [Crossref]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, et al. (2014) Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514: 628-632 [Crossref]
- Inman KS, Francis AA, Murray NR (2014) Complex role for the immune system in initiation and progression of pancreatic cancer. World J Gastroenterol 20: 11160-11181. [Crossref]
- Clark CE, Beatty GL, Vonderheide RH (2009) Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Lett 279: 1–7. [Crossref]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, et al. (2012) EMT and dissemination precede pancreatic tumor formation. Cell 148: 349-361. [Crossref]
- Xie D, Xie K1 (2015) Pancreatic cancer stromal biology and therapy. Genes Dis 2: 133-143. [Crossref]
- Yadav D, Lowenfels AB (2013) The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 144: 1252-1261. [Crossref]
- Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M (2013) Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology 144: 1199-1209. [Crossref]
- Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, et al. (2016) Bruton’s Tyrosine Kinase (BTK)-dependent immune cell crosstalk drives pancreas cancer. Cancer Discov 6: 270-285. [Crossref]
- Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, et al. (2011) STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res 71: 5020-5029. [Crossref]
- Evans A, Costello E (2012) The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front Physiol 3: 270. [Crossref]
- Ademmer K, Ebert M, Müller-Ostermeyer F, Friess H, Büchler MW, et al. (1998) Effector T lymphocyte subsets in human pancreatic cancer: detection of CD8+CD18+ cells and CD8+CD103+ cells by multi-epitope imaging.Clin Exp Immunol 112: 21-26. [Crossref]
- Hiraoka N, Onozato K, Kosuge T, Hirohashi S (2006) Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res 12: 5423-5434. [Crossref]
- Husain Z, Huang Y, Seth P, Sukhatme VP (2013) Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol 19:1486-1495. [Crossref]
- Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M (2013) Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology 144: 1199-1209. [Crossref]
- Yanagimoto H, Takai S, Satoi S, Toyokawa H, Takahashi K, et al. (2005) Impaired function of circulating dendritic cells in patients with pancreatic cancer. Clin Immunol 114: 52-60. [Crossref]
- Ryschich E, Nötzel T, Hinz U, Autschbach F, Ferguson J, et al. (2005) Control of T-cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin Cancer Res 1: 498–504. [Crossref]
- von Bernstorff W, Spanjaard RA, Chan AK, Lockhart DC, Sadanaga N, et al. (1999) Pancreatic cancer cells can evade immune surveillance via nonfunctional Fas (APO-1/CD95) receptors and aberrant expression of functional Fas ligand. Syrgery 125: 73-84. [Crossref]
- Suzuki N, Maeda Y, Tanaka S, Hida N, Mine T, et al. (2002) Detection of peptide-specific cytotoxic T-lymphocyte precursors used for specific immunotherapy of pancreatic cancer. Int J Cancer 98: 45–50.
- Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, et al. (2004) CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 28: 26–31. [Crossref]
- Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, et al. (1998) Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 43: 128–133. [Crossref]
- Bachem MG, Zhou S, Buck K, Schneiderhan W, Siech M (2008) Pancreatic stellate cells--role in pancreas cancer. Langenbecks Arch Surg 393: 891-900. [Crossref]
- Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, et al. (2004) Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells.Pancreas 29:179e87. [Crossref]
- Apte MV, Wilson JS, Lugea A, Pandol SJ (2013) A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 144: 1210-1219. [Crossref]
- Apte MV, Xu Z, Pothula S, Goldstein D, Pirola RC, et al. (2015) Pancreatic cancer: The microenvironment needs attention too! Pancreatology 15: S32-38. [Crossref]
- Fukushima N, Kikuchi Y, Nishiyama T, Kudo A, Fukayama M (2008) Periostin deposition in the stroma of invasive and intraductal neoplasms of the pancreas. Mod Pathol 2: 1044–1053. [Crossref]
- Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, et al. (2013) Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res 73: 3007-3018. [Crossref]
- Mace TA, Bloomston M, Lesinski GB (2013) Pancreatic cancer associated stellate cells: A viable target for reducing immunosuppression in the tumor microenvironment. Oncoimmunology 2: e24891. [Crossref]
- Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, et al. (2013) Activated pancreatic stellate cells sequester CD8+ T-cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 145: 1121-1132. [Crossref]
- Gao Z, Wang X, Wu K, Zhao Y, Hu G (2010) Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/CXCR4 axis. Pancreatology 10: 186–193. [Crossref]
- Xu Z (2010) Role of pancreatic stellate cells in pancreatic ductal adenocarcinoma metastasis. Am J Pathol 177:2585–2596.
- Tien YW, Wu YM, Lin WC, lee HS, Le HS (2009) Pancreatic carcinoma cells stimulate proliferation and matrix synthesis of hepatic stellate cells. J Hepatol 5: 307–314.
2021 Copyright OAT. All rights reserv
- Kordes C, Sawitza I, Häussinger D (2009) Hepatic and pancreatic stellate cells in focus. Biol Chem 390: 1003-1012. [Crossref]
- Li X, Wang Z, Ma Q, Xu Q, Liu H, et al. (2014) Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin Cancer Res 20: 4326-4338. [Crossref]
- Preston M, Sherman LS (2011) Neural stem cell niches: roles for the hyaluronan-based extracellular matrix. Front Biosci 3:1165–1179. [Crossref]
- Schuller HM (2008) Neurotransmission and cancer: implications for prevention and therapy. Anticancer Drugs 19: 655-671. [Crossref]
- Zhu Z, Kleeff J, Kayed H, Wang L, Korc M, et al. (2002) Nerve growth factor and enhancement of proliferation, invasion, and tumorigenicity of pancreatic cancer cells. Mol Carcinog 35: 138–147. [Crossref]
- Lunardi S, Muschel RJ, Brunner TB (2014) The stromal compartments in pancreatic cancer: are there any therapeutic targets? Cancer Lett 343: 147-55.
- Ottaviano AJ, Sun L, Ananthanarayanan V, Munshi HG (2006) Extracellular matrix-mediated membrane-type 1 matrix metalloproteinase expression in pancreatic ductal cells is regulated by transforming growth factor-ß1. Cancer Res 66: 7032–7040. [Crossref]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD (2012) Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2: 418-429. [Crossref]
- Seo Y, Baba H, Fukuda T, Takashima M, Sugimachi K (2000) High expression of vascular endothelial growth factor is associated with liver metastasis and an poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer 8: 2239–2245. [Crossref]
- Wang HX, Chen QK (2003) [Expression and significance of cyclooxygenase-2 in human pancreatic carcinomas]. Ai Zheng 22: 649-652. [Crossref]
- Fukahi K, Fukasawa M, Neufeld G, Itakura J, Korc M (2004) Aberrant expression of neuropilin-1 and -2 in human pancreatic cancer cells. Clin Cancer Res 10: 581-590. [Crossref]
- Erkan M (2009) Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 1:497–508.
- Mclntyre CA, Winter JM (2015) Diagnostic Evaluation and Staging of Pancreatic Ductal Adenocarcinoma. Semin Oncol 42: 19-27. [Crossref]
- Li D (2012) Diabetes and pancreatic cancer. Mol Carcinog 51: 64-74. [Crossref]
- Mayr M, Schmid RM (2010) Pancreatic cancer and depression: myth and truth. BMC Cancer 10: 569. [Crossref]
- Apple F, Benson P, Preese L, Eastep S, Bilodeau L, et al. (1991) Lipase and pancreatic amylase activities in tissues and in patients with hyperamylasemia. Am J Clin Pathol 96: 610-614. [Crossref]
- Winter JM, Yeo CJ, Brody JR (2013) Diagnostic, prognostic, and predictive biomarkers in pancreatic cancer. J Surg Oncol 107: 15-22. [Crossref]
- Al-Hawary MM, Francis IR (2013) Pancreatic ductal adenocarcinoma staging. Cancer Imaging 13: 360-364. [Crossref]
- Edge SB, Compton CC (2010) The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 17: 1471-1474. [Crossref]
- Tempero MA, Arnoletti JP, Behrman SW, Ben-Josef E, Benson AB 3rd, et al. (2012) Pancreatic Adenocarcinoma, version 2.2012: featured updates to the NCCN Guidelines. J Natl Compr Canc Netw 10: 703-713. [Crossref]
- Vauthey JN, Dixon E (2009) AHPBA/SSO/SSAT Consensus Conference on Resectable and Borderline Resectable Pancreatic Cancer: rationale and overview of the conference. Ann Surg Oncol 16: 1725-1726. [Crossref]
- Yachida S, Iacobuzio-Donahue CA (2009) The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med 133: 413-422. [Crossref]
- Hidalgo M, Cascinu S, Kleeff J, Labianca R, Löhr JM, et al. (2015) Addressing the challenges of pancreatic cancer: future directions for improving outcomes. Pancreatology 15: 8-18. [Crossref]
- Sclafani F, Iyer R, Cunningham D, Starling (2015) Management of metastatic pancreatic cancer: Current treatment options and potential new therapeutic targets. Crit Rev Oncol Hematol 95: 318-336. [Crossref]
- Neoptolemos JP, Dunn JA, Stocken DD, Almond J, Link K, et al. (2001) Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomized controlled trial. Lancet 358: 1576–1585. [Crossref]
- [No authors listed] (1987) GITSG. Further evidence of effective adjuvant combined radiation and chemotherapy following curative resection of pancreatic cancer. Gastrointestinal Tumor Study Group. Cancer 59: 2006-2010. [Crossref]
- Klinkenbijl JH, Jeekel J, Sahmoud T, van Pel R, Couvreur ML, et al. (1999) Adjuvant radiotherapy and 5-fluorouracil after curative resection of cancer of the pancras and periampullary region: Phase III trial of the EORTC gastrointestinal tract cancer cooperative group. Ann Surg 230: 776–782. [Crossref]
- Neoptolemos JP, Stocken DD, Dunn JA, Almond J, Beger HG, et al. (2001) The influence of resection margins on survival for patients with pancreatic cancer treated by adjuvant chemoradiation and/or chemotherapy in the ESPAC-1 randomized controlled trial. Ann Surg 234: 758–768. [Crossref]
- Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, et al. (2004) A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 350: 1200-1210. [Crossref]
- Oettle H (2013) Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA 310: 1473-1481. [Crossref]
- Neoptolemos JP, Stocken DD, Bassi C, Ghaneh P, Cunningham D, et al. (2010) Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA 304: 1073-1081. [Crossref]
- Regine W (2006) A phase III Intergroup trial (RTOG 97-04) of adjuvant pre and post chemoradiation (CRT) 5-FU vs. gemcitabine (G) for resected pancreatic adenocarcinoma. Int J Radiat Oncol Biol Phys 66: 23–24.
- Katz MH1, Pisters PW, Evans DB, Sun CC, Lee JE, et al. (2008) Borderline resectable pancreatic cancer: the importance of this emerging stage of disease. J Am Coll Surg 206: 833–848. [Crossref]
- Abrams RA1, Lowy AM, O'Reilly EM, Wolff RA, Picozzi VJ, et al. (2009) Combined modality treatment of resectable and borderline resectable pancreas cancer: expert consensus statement. Ann Surg Oncol 16: 1751-1756. [Crossref]
- Sclafani F, Iyer R, Cunningham D, Starling N (2015) Management of metastatic pancreatic cancer: Current treatment options and potential new therapeutic targets. Crit Rev Oncol Hematol 95: 318-336. [Crossref]
- Kim HW, Lee JC, Paik KH, Lee YS, Hwang JH, et al. (2015) Initial Metastatic Site as a Prognostic Factor in Patients With Stage IV Pancreatic Ductal Adenocarcinoma. Medicine (Baltimore) 94: e1012. [Crossref]
- Huguet F, Mukherjee S2, Javle M3 (2014) Locally advanced pancreatic cancer: the role of definitive chemoradiotherapy. Clin Oncol (R Coll Radiol) 26: 560-568. [Crossref]
- Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, et al. (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15: 2406-2413. [Crossref]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, et al. (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25:1960–1966. [Crossref]
- Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, et al. (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364: 1817-1825. [Crossref]
- Ho MY, Kennecke HF, Renouf DJ, Cheung WY, Lim HJ, et al. (2015) Defining eligibility of FOLFIRINOX for the first-line metastatic pancreatic adenocarcinoma (MPC) in the province of the British Columbia: a population based retrospective study. Am J Clin Oncol [Crossref]
- Mahaseth H, Brutcher E, Kauh J, Hawk N, Kim S (2013) Modified FOLFIRINOX regimen with improved safety and maintained efficacy in pancreatic adenocarcinoma. Pancreas 42: 1311-1315. [Crossref]
- Gunturu KS, Yao X, Cong X, Thumar JR, Hochster HS, et al. (2013) FOLFIRINOX for locally advanced and metastatic pancreatic cancer: single institution retrospective review of efficacy and toxicity. Med Oncol 30: 361. [Crossref]
- Blazer M, Wu C, Goldberg RM, Phillips G, Schmidt C, et al. (2015) Neoadjuvant Modified (m) FOLFIRINOX for Locally Advanced Unresectable (LAPC) and Borderline Resectable (BRPC) Adenocarcinoma of the Pancreas. Ann Surg Oncol 22: 1153-1159. [Crossref]
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, et al. (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369: 1691-1703. [Crossref]
- Al-Hajeili M, Azmi AS2, Choi M3 (2014) Nab-paclitaxel: potential for the treatment of advanced pancreatic cancer. Onco Targets Ther 7: 187-192. [Crossref]
- Pelzer U, Schwaner I, Stieler J, Adler M, Seraphin J, et al. (2011) Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III study from the German CONKO-study group. Eur J Cancer 47: 1676–1681. [Crossref]
- Berger AK, Weber TF, Jäger D, Springfeld C (2013) Successful treatment with nab-paclitaxel and gemcitabine after FOLFIRINOX failure in a patient with metastasized pancreatic adenocarcinoma. Onkologie. 36, 763-765. [Crossref]
- Portal A, Pernot S, Tougeron D, Arbaud C, Bidault AT, et al. (2015) Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: an AGEO prospective multicentre cohort. Br J Cancer 113, 989-995. [Crossref]
- Zhang Y, Hochster H, Stein S, Lacy J (2015) Gemcitabine plus nab-paclitaxel for advanced pancreatic cancer after first-line FOLFIRINOX: single institution retrospective review of efficacy and toxicity. Exp Hematol Oncol 4:29.[Crossref]
- da Rocha Lino A, Abrahão CM, Brandão RM, Gomes JR, Ferrian AM, et al. (2015) Role of gemcitabine as second-line therapy after progression on FOLFIRINOX in advanced pancreatic cancer: a retrospective analysis. J Gastrointest Oncol 6: 511-51. [Crossref]
- Wang-Gillam A, Li CP2, Bodoky G, Dean A, Shan YS, et al. (2016) Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. The Lancet 387: 545-557. [Crossref]
- Reni M, Cereda S, Milella M, Novarino A, Passardi A, et al. (2013) Maintenance sunitinib or observation in metastatic pancreatic adenocarcinoma: a phase II randomised trial. Eur J Cancer 49: 3609-3615. [Crossref]
- Birk D, Beger HG (2001) Neoadjuvant, adjuvant, and palliative treatment of pancreatic cancer. Curr Gastroenterol Rep 3: 129–135. [Crossref]
- De La Cruz MS, Young AP2, Ruffin MT2 (2014) Diagnosis and management of pancreatic cancer. Am Fam Physician 89: 626-632. [Crossref]
- Sikkens EC, Cahen DL, Kuipers EJ, Bruno MJ (2010) Pancreatic enzyme replacement therapy in chronic pancreatitis. Best Pract Res Clin Gastroenterol 24: 337-347. [Crossref]
- Costamagna G, Alevras P, Palladino F, Rainoldi F, Mutignani M, et al. (1999) Endoscopic pancreatic stenting in pancreatic cancer. Can J Gastroenterol 13: 481-487. [Crossref]
- Stark, A. & Hines, O.J. Endoscopic and operative palliation strategies for pancreatic ductal adenocarcinoma. Semin. Oncol. 42, 163-176 (2015).
- Zhong W, Yu Z, Zeng JX, Lin Y, Yu T, et al. (2014) Celiac plexus block for treatment of pain associated with pancreatic cancer: a meta-analysis. Pain Pract 14: 43-51. [Crossref]
- Lili LN, Matyunina LV, Walker LD, Daneker GW, McDonald JF (2014) Evidence for the importance of personalized molecular profiling in pancreatic cancer. Pancreas 43: 198-211. [Crossref]
- Yachida S (2010) Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467: 1114–1117.
- Sheikh R, Walsh N, Clynes M, O'Connor R, McDermott R (2010) Challenges of drug resistance in the management of pancreatic cancer. Expert Rev Anticancer Ther 10: 1647-1661. [Crossref]
- Koay EJ1, Baio FE, Ondari A, Truty MJ, Cristini V, et al. (2014) Intra-tumoral heterogeneity of gemcitabine delivery and mass transport in human pancreatic cancer. Phys Biol 11:0650002. [Crossref]
- Nakahira (2007) Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int. J. Cancer 120: 1355–1363 [Crossref]
- Duxbury, M.S., Ito, H., Zinner, M.J., Ashley, S.W., Whang, E.E. (2004) RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine. Oncogene. 23: 1539–1548 [Crossref]
- Galmarini CM, Clarke ML, Falette N, Puisieux A, Mackey JR, et al. (2002) Expression of a non-functional p53 affects the sensitivity of cancer cells to gemcitabine. Int J Cancer 97: 439-445. [Crossref]
- Bold, RJ, Chandra J, McConkey DJ (1999) Gemcitabine-induced programmed cell death (apoptosis) of human pancreatic carcinoma is determined by Bcl 2 content. Ann Surg Oncol 6: 279–285 [Crossref]
- Ali (2010) Gemcitabine Sensitivity Can Be Induced in Pancreatic Cancer Cells through Modulation of miR-200 and miR-21 Expression by Curcumin or Its Analogue CDF. Cancer Res 70: 3606–3617 [Crossref]
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE (2004) Inhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells. Clin Cancer Res 10: 2307–2318 [Crossref]
- Arlt (2003) A Role of NF kB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 22: 3243–3251 [Crossref]
- Kasuya K (2011) Hypoxia-inducible factor-1a expression and gemcitabine chemotherapy for pancreatic cancer. Oncol Rep 26: 1399-1406 [Crossref]
- Wang Z (2009) Acquisition of epithelial–mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 69: 2400–2407. [Crossref]
- Ischenko I (2008) Inhibition of Src tyrosine kinase reverts chemoresistance toward 5-fluorouracil in human pancreatic carcinoma cells: an involvement of epidermal growth factor receptor signaling. Oncogene 27: 7212–7222 [Crossref]
- Shi X, Liu S, Kleeff J, Friess H, Büchler MW (2002) Acquired resistance of pancreatic cancer cells towards 5-fluorouracil and gemcitabine is associated with altered expression of apoptosis-regulating genes. Oncology 62: 354–362. [Crossref]
- Hagmann W, Jesnowski R, Faissner R, Guo C, Löhr JM (2009) Atp-binding cassette c transporters in human pancreatic carcinoma cell lines. Upregulation in 5-fluorouracil-resistant cells. Pancreatology. 9, 136–144. [Crossref]
- Nambaru PK, Hübner T, Köck K, Mews S, Grube M, et al. (2010) Drug efflux transporter multidrug resistance-associated protein 5 affects sensitivity of pancreatic cancer cell lines to the nucleoside anticancer drug 5-fluorouracil. Drug Metab Dispos 39: 132–139. [Crossref]
- Rachakonda PS, Bauer AS, Xie H, Campa D, Rizzato C, et al. (2013) Somatic mutations in exocrine pancreatic tumors: association with patient survival. PLoS One 8: e60870. [Crossref]
- Cohen SJ (2003) Phase II and pharmacodynamic study of the farnesyltransferase inhibitor R115777 as initial therapy in patients with metastatic pancreatic adenocarcinoma. J Clin Oncol 2: 1301-6.
- Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, et al. (2004) Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol 22: 1430-1438. [Crossref]
- Lerner EC, Zhang TT, Knowles DB, Qian Y, Hamilton AD, et al. (1997) Inhibition of the prenylation of K-Ras, but not H- or N-Ras, is highly resistant to CAAX peptidomimetics and requires both a farnesyltransferase and a geranylgeranyltransferase I inhibitor in human tumor cell lines. Oncogene. 15, 1283–1288. [Crossref]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, et al. (2005) KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2: e17. [Crossref]
- Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, et al. (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359: 1757–1765. [Crossref]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039–1043. [Crossref]
- Yamasaki F, Johansen MJ, Zhang D, Krishnamurthy S, Felix E, et al. (2007) Acquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res. 67, 5779–5788. [Crossref]
- Chen HX, Sharon E (2013) IGF-1R as an anti-cancer target--trials and tribulations. Chin J Cancer 32: 242-252. [Crossref]
- Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. (2013) Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS One 8: e57289. [Crossref]
- Michl P, Gress TM (2012) Improving drug delivery to pancreatic cancer: Breaching the stromal fortress by targeting hyaluronic acid. Gut 6:1377–1379. [Crossref]
- Olive KP1, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, et al. (2009) Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324: 1457–1461. [Crossref]
- Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, et al. (2014) Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res 20: 5937-5945. [Crossref]
- De Jesus-Acosta A (2014) A Phase II study of vismodegib, a hedgehog (Hh) pathway inhibitor, combined with gemcitabine and nab-paclitaxel (nab-P) in patients with untreated metastatic pancreatic ductal adenocarcinoma (PDA). J Clin Oncol 32(Suppl): 257.
- Catenacci, D.V.T. et al. (2015) Randomized phase Ib/ II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol 33: 4284-4292. [Crossref]
- Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, et al. (2014) Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A 11: 3091-3100. [Crossref]
- Grützmann R, Boriss H, Ammerpohl O, Lüttges J, Kalthoff H, et al. (2005) Meta-analysis of microarray data on pancreatic cancer defines a set of commonly dysregulated genes. Oncogene 24: 5079-5088. [Crossref]
- Miyamoto H, Murakami T, Tsuchida K, Sugino H, Miyake H, et al. (2004) Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 28: 38–44. [Crossref]
- Toste PA, Nguyen AH, Kadera BE, Duong M, Wu N, et al. (2016) Chemotherapy-induced inflammatory gene signature and pro-tumorigenic phenotype in pancreatic CAFs via stress-associated MAPK. Mol Cancer Res [Crossref]
- Morimoto M, Matsuo Y2, Koide S, Tsuboi K, Shamoto T, et al. (2016) Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: effect of CXCR4 antagonists. BMC Cancer16: 305. [Crossref]
- Royal RE, Levy C, Turner K, Mathur A, Hughes M et al. (2010) Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 33: 828-833. [Crossref]
- Pothula SP, Xu Z, Goldstein D, Biankin AV, Pirola RC, et al. (2016) Hepatocyte growth factor inhibition: a novel therapeutic approach in pancreatic cancer. BJ Cancer 114:269-280. [Crossref]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, et al. (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444: 756-760. [Crossref]
- Venezia TA, Merchant AA, Ramos CA, Whitehouse NL, Young AS, et al. (2004) Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol 2: e301. [Crossref]
- Kim MP, Fleming JB, Wang H, Abbruzzese JL, Choi W, et al. (2011) ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PloS One 6: e20636. [Crossref]
- O’Reilly EM (2015) BRCA-mutated pancreas adenocarcinoma: emerging therapeutic implications. AACR Special Conference on Pancreatic Cancer. Meeting Abstract IA28. Cancer Res 75.
- O’Reilly EM (2014) Phase IB trial of cisplatin, gemcitabine, and veliparib in patients with known or potential BRCA or PALB2- mutated pancreas adenocarcinoma. J Clin Oncol 32:5s.
- Venkatasubbarao K, Peterson L, Zhao S, Hill P, Cao L, et al. (2013) Inhibiting signal transducer and activator of transcription-3 increases response to gemcitabine and delays progression of pancreatic cancer. Mol Cancer 12:104. [Crossref]
- Slusarczyk M, Lopez MH, Balzarini J, Mason M, Jiang WG, et al. (2014) Application of ProTide technology to gemcitabine: a successful approach to overcome the key cancer resisatnce mechanisms leads to a new agent (NUC-1) in clinical development. J Med Chem 57: 1531-1542. [Crossref]
- Blagden S (2015) Final results from the first human phase I/II study of NUC-1031 in patients with solid tumours. ASCO. Abstract no: 2514.
- Gilabert M, Calvo E, Airoldi A, Hamidi T, Moutardier V, et al. (2014) Pancreatic cancer-induced cachexia is Jak2-dependent in mice. J Cell Physiol 229: 1437-1443. [Crossref]
- Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, et al. (2015) Randomized, Double-Blind, Phase II Study of Ruxolitinib or Placebo in Combination With Capecitabine in Patients With Metastatic Pancreatic Cancer for Whom Therapy With Gemcitabine Has Failed. J Clin Oncol 33: 4039-4047. [Crossref]
- Lee HS, Lee SH, Lee HJ, Chung MJ, Park JY, et al. (2016) Statin Use and Its Impact on Survival in Pancreatic Cancer Patients. Medicine (Baltimore) 95: e3607. [Crossref]
- Sadeghi N, Abbruzzese JL, Yeung SC, Hassan M, Li D (2012) Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin Cancer Res 18: 2905-2912. [Crossref]
- Zhang P, Li H, Tan X, Chen L, Wang S (2013) Association of metformin use with cancer incidence and mortality: a meta-analysis. Cancer Epidemiol 37: 207-218. [Crossref]
- Fasih A, Elbaz HA, Hüttemann M, Konski AA, Zielske SP (2014) Radiosensitization of pancreatic cancer cells by metformin through the AMPK pathway. Radiat Res 182: 50-59. [Crossref]
- Karnevi E, Said K, Andersson R, Rosendahl AH (2012) Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer 13: 235. [Crossref]
- Wilmink J (2014) A phase II randomized, placebo controlled study to evaluate the efficacy of the combination of gemcitabine, erlotinib, and metformin in patients with locally advanced or metastatic pancreatic cancer. ASCO Annual Meeting. J Clin Oncol. Chicago.
- Braghiroli MI, de Celis Ferrari AC, Pfiffer TE, Alex AK, Nebuloni D, et al. (2015) Phase II trial of metformin and paclitaxel for patients with gemcitabine-refractory advanced adenocarcinoma of the pancreas. Ecancer 9: 563. [Crossref]
- Yabuuchi S, Pai SG, Campbell NR, de Wilde RF, De Oliveira E, et al. (2013) Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett 335: 41-51. [Crossref]
- Mueller MT (2009)Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology 137: 1102-1113. [Crossref]
- Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, et al. (2015) MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab 22: 590-605. [Crossref]
- Zhao L, Zhao Y, Schwarz B, Mysliwietz J, Hartig R, et al. (2016) Verapamil inhibits tumour progression of chemotherapy-resistant pancreatic cancer side population cells. Int J Oncol 49: 99-110. [Crossref]
- Yardley DA (2013) nab-Paclitaxel mechanisms of action and delivery. J Control Release 170: 365-372. [Crossref]
- Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, et al. (2011) Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 29: 4548-4554. [Crossref]
- Alvarez R, Musteanu M, Garcia-Garcia E, Lopez-Casas PP, Megias D, et al. (2013) Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br J Cancer 109: 926-933. [Crossref]
- Gonzalez-Villasana V, Rodriguez-Aguayo C, Arumugam T, Cruz-Monserrate Z, Fuentes-Mattei E, et al. (2014) Bisphosphonates Inhibit Stellate Cell Activity and Enhance Antitumor Effects of Nanoparticle Albumin Bound-Paclitaxel in Pancreatic Ductal Adenocarcinoma Mol. Cancer Ther 13: 2583–2594. [Crossref]
- Pothula SP, Xu Z, Goldstein D, Biankin AV, Pirola RC, et al. (2016) Hepatocyte growth factor inhibition: a novel therapeutic approach in pancreatic cancer. Br J Cancer 114: 269-280. [Crossref]
- Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, et al. (2016) Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Discov 6: 256-269. [Crossref]
- Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, et al. (2016) Bruton’s Tyrosine Kinase (BTK)-dependent immune cell crosstalk drives pancreas cancer. Cancer Discov 6: 270-285. [Crossref]
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, et al. (2011) CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 33: 1612-1616. [Crossref]
- Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, et al. (2013) A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma.Clin Cancer Res 19: 6286-6295. [Crossref]
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, et al. (2013) Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 110: 20212-20217. [Crossref]
- Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S, et al. (2012) A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: Phase I studies of safety and immune induction. Clin Cancer Res 18: 858-868. [Crossref]
- Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, et al. (2015) Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol 33:1325-1333. [Crossref]
- McEwan C, Owen J, Stride E, Fowley C, Nesbitt H, et al. (2015) Oxygen carrying microbubbles for enhanced sonodynamic therapy of hypoxic tumours. J Control Release 203: 51-56. [Crossref]
- McEwan C, Kamila S, Owen J, Nesbitt H, Callan B, et al. Combined sonodynamic and antimetabolite therapy for the improved treatment of pancreatic cancer using oxygen loaded microbubbles as a delivery vehicle. Biomaterials 80: 20-32 (2016). [Crossref]
- Mullard A (2014) Learning from exceptional drug responders. Nat Rev Drug Discov 13: 401-402. [Crossref]
- Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, et al. (2012) Genome sequencing identifies a basis for everolimus sensitivity. Science 338: 221. [Crossref]
- Subbiah V, Naing A, Brown RE, Chen H, Doyle L, et al. (2011) Targeted morphoproteomic profiling of Ewing’s sarcoma treated with insulin-like growth factor 1 receptor (IGF1R) inhibitors: response/resistance signatures. PLoS ONE 6: e18424. [Crossref]
- Chue BM (2009) Five-year survival of metastatic pancreatic carcinoma: a study of courage and hope. Gastrointest Cancer Res 3: 208-211. [Crossref]
- Klimant E, Markman M, Albu DM (2013) Clinically Meaningful Responses to Sequential Gemcitabine-Based Chemotherapy Regimens in a Patient with Metastatic Pancreatic Cancer. Case Rep Oncol 6: 72–77. [Crossref]
- Rios Perez MV, Dai B, Koay EJ, Wolff RA, Fleming JB (2015) Regression of Stage IV Pancreatic Cancer to Curative Surgery and Introduction of a Novel Ex-Vivo Chemosensitivity Assay. Cureus 7:e423. [Crossref]
- Cen P, Ni X, Yang J, Graham DY, Li M (2012) Circulating tumor cells in the diagnosis and management of pancreatic cancer. Biochim Biophy Acta 1826: 350-356. [Crossref]
- Poruk KE, Valero V 3rd, Saunders T, Blackford AL, Griffin JF, et al. (2016) Circulating tumor cells phenotype predicts recurrence and survival in pancreatic adenocarcinoma. Ann Surg 264:1073-1081. [Crossref]
- Garcea G, Ladwa N, Neal CP, Metcalfe MS, Dennison AR, et al. (2011) Preoperative neutrophil-to-lymphocyte ratio (NLR) is associated with reduced disease-free survival following curative resection of pancreatic adenocarcinoma. World J Surg 35: 868–872. [Crossref]
- Radon TP, Massat NJ, Jones R, Alrawashdeh W, Dumartin L, et al. (2015) Identification of a three-biomarker panel in urine for early detection of pancreatic adenocarcinoma. Clin Cancer Res 2: 3512. [Crossref]
- Ohtsubo K, Watanabe H, Yao F, Okada G, Mouri H, et al. Preproenkephalin hypermethylation in the pure pancreatic juice compared with p53 mutation in the diagnosis of pancreatic carcinoma. J Gastroenterol 4: 791-797 (2006). [Crossref]
- Stratford JK, Bentrem DJ, Anderson JM, Fan C, Volmar KA, et al. (2010) A six-gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Med 7: e1000307. [Crossref]
- Manuyakorn A, Paulus R, Farrell J, Dawson NA, Tze S, et al. (2010) Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J Clin Oncol 28: 1358–1365. [Crossref]
- Moravec R, Divi R, Verma M (2017) Detecting circulating tumor material and digital pathology imaging during pancreatic cancer progression. World J Gastrointest Oncol 9: 235-250. [Crossref]
- Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, et al. (2015) Glypican1 identifies cancer exosomes and facilitates early detection of cancer. Nature 523: 177-182. [Crossref]
- Ghazaly EA (2015) Validating the RNAscope for molecular profiling of key biomarkers associated with gemcitabine resistance. Cancer Res 75: 3390.
- Orlandi A, Calegari MA, Martini M, Cocomazzi A, Bagalà C, et al. (2016) Gemcitabine versus FOLFIRINOX in patients with advanced pancreatic adenocarcinoma hENT1-positive: everything was not too bad back when everything seemed worse. Clin Transl Oncol 18: 988-995. [Crossref]
- Hudis CA (2007) Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 357: 39-51. [Crossref]
- Wang JP, Wu CY, Yeh YC, Shyr YM, Wu YY, et al. (2015) Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: a randomized, open-label, propective trial. Oncotarget 6: 18162-73. [Crossref]
- Golan T, Atias D, Barshack I, Avivi C, Goldstein RS, et al. (2014) Ascites-derived pancreatic ductal adenocarcinoma primary cell cultures as a platform for personalised medicine. Br J Cancer 110: 2269-2276. [Crossref]
- Zhang HC, Kuo CJ (2015) Personalizing pancreatic cancer organoids with hPSCs. Nat Med 21: 1249-1251. [Crossref]
- Keshet Y, Schiff E, Samuels N, Ben-Arye E (2015) Giving voice to cancer patients: assessing non-specific effects of an integrative oncology therapeutic program via short patient narratives. Psychooncology 24: 169-174. [Crossref]
- Sagar SM, Leis AM (2008) Integrative oncology: a Canadian and international perspective. Curr Oncol 15 Suppl 2: s71-73. [Crossref]
- Deng G, Cassileth B (2013) Complementary or alternative medicine in cancer care-myths and realities. Nat Rev Clin Oncol 10: 656-664. [Crossref]
- Marchand L (2014) Integrative and complementary therapies for patients with advanced cancer. Ann Palliat Med 3: 160-171. [Crossref]
- Campbell KL, McTiernan A (2007) Exercise and biomarkers for cancer prevention studies. J Nutr 137: 161S-169S. [Crossref]
- World Cancer Research Fund /American Institute for Cancer Research (2007) Food nutrition, physical activity, and the prevention of cancer: a global perspective. Washington, DC: AICR.
- Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140: 883-899. [Crossref]
- Neuzillet C, Vergnault M, Bonnetain F, Hammel P (2015) Rationale and design of the Adapted Physical Activity in advanced Pancreatic Cancer patients (APACaP) GERCOR (Groupe Coopérateur Multidisciplinaire en Oncologie) trial: study protocol for a randomized controlled trial. Trials 16: 454. [Crossref]
- Frenkel M, Sierpina V, Sapire K (2015) Effects of complementary and integrative medicine on cancer survivorship. Curr Oncol Rep 17: 445. [Crossref]
- Sagar SM, Lawenda BD (2009) The role of integrative oncology in a tertiary prevention survivorship program. Prev Med 49: 93-98. [Crossref]
- Yang EV, Glaser R (2003) Stress-induced immunomodulation: implications for tumorigenesis. Brain Behav Immun 17: 37–S40. [Crossref]
- Chan C, Lin HJ, Lin J (2008) Stress-associated hormone, norepinephrine, increases proliferation and IL-6 levels of human pancreatic duct epithelial cells and can be inhibited by the dietary agent, sulforaphane. Int J Oncol 33: 415-419. [Crossref]
- Genkinger JM, Wang M, Li R, Albanes D, Anderson KE, et al. (2014) Dairy products and pancreatic cancer risk: a pooled analysis of 14 cohort studies. Ann Oncol 25:1106-1115. [Crossref]
- Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN (2010) Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research. Carcinogenesis 3: 83–89. [Crossref]
- Lashinger LM, Malone LM, Brown GW, Daniels EA, Goldberg JA, et al. (2011) Rapamycin partially mimics the anticancer effects of calorie restriction in a murine model of pancreatic cancer. Cancer Prev Res (Phila) 4: 1041-51. [Crossref]
- Harvey AE, Lashinger LM, Hays D, Harrison LM, Lewis K, et al. (2014) Calorie restriction decreases murine and human pancreatic tumor cell growth, nuclear factor-kB activation, and inflammation related gene expression in an insulin-like growth Factor-12 dependent manner. PLOS ONE 9: E94151. [Crossref]
- Lanza-Jacoby S, Yan G, Radice G, LePhong C, Baliff J, et al. (2013) Calorie restriction delays the progression of lesions to pancreatic cancer in the LSL-KrasG12D; Pdx-1/Cre mouse model of pancreatic cancer. Exp Biol Med (Maywood) 238: 787-97. [Crossref]
- Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, et al. (2014) Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab 2:18. [Crossref]
- Uomo G, Gallucci F, Rabitti PG (2006) Anorexia-cachexia syndrome in pancreatic cancer: recent development in research and management. JOP 7: 157-62. [Crossref]
- Hashim SA, VanItallie TB1 (2014) Ketone body therapy: from the ketogenic diet to the oral administration of ketone ester. J Lipid Res 55: 1818-1826. [Crossref]
- Tang SN, Fu J, Shankar S, Srivastava RK (2012) EGCG enhances the therapeutic potential of gemcitabine and CP690550 by inhibiting STAT3 signaling pathway in human pancreatic cancer. PLoS One 7: e31067. [Crossref]
- Kanai M, Yoshimura K, Asada M, Imaizumi A, Suzuki C, et al. (2011) A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother Pharmacol 68: 157-164. [Crossref]
- Li Y, VandenBoom TG 2nd, Kong D, Wang Z, Ali S, et al. (2009) Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res 69: 6704-6712. [Crossref]
- Harikumar KB, Kunnumakkara AB, Sethi G, Diagaradjane P, Anand P, et al. (2010) Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. Int J Cancer 127: 257-268. [Crossref]
- Arshad A, Isherwood J, Mann C, Cooke J, Pollard C, et al. (2015) Intravenous ɷ-3 Fatty Acids Plus Gemcitabine: Potential to Improve Response and Quality of Life in Advanced Pancreatic Cancer. JPEN J Parenter Enteral Nutr [Crossref]
- Neoptolemos JP, Stocken DD, Bassi C, Ghaneh P, Cunningham D, et al. (2010) Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA 304: 1073-1081. [Crossref]
- Cheng X, Kim JY, Ghafoory S, Duvaci T, Rafiee R, et al. (2016) Methylisoindigo preferentially kills cancer stem cells by interfering cell metabolism via inhibition of LKB1 and activation of AMPK in PDACs. Mol Oncol 10: 806-824. [Crossref]
- Zhang Y, Chen AY, Li M, Chen C, Yao Q (2008) Ginkgo biloba extract kaempferol inhibits cell proliferation and induces apoptosis in pancreatic cancer cells. J Surg Res 148: 17–23. [Crossref]
- Ferguson LR, Chen H, Collins AR, Connell M, Damia G, et al. (2015) Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin Cancer Biol 35, 5-24. [Crossref]
- Banerjee S, Zhang Y, Wang Z, Che M, Chiao PJ, et al. (2006) In vitro and in vivo molecular evidence of genistein action in augmenting the efficacy of cisplatin in pancreatic cancer. Int J Cancer 120: 906–917. [Crossref]
- Bhattacharjee Y, Zhou Y, Yen TJ (2014) A synthetic lethal screen identifies the Vitamin D receptor as a novel gemcitabine sensitizer in pancreatic cancer cells. Cell Cycle 13: 3838-3856. [Crossref]
- Persons KS, Eddy VJ, Chadid S, Deoliveira R, Saha AK, et al. (2010) Anti-growth effect of ,25-dihydroxyvitamin D3-3-bromoacetate alone or in combination with 5-amino-imidazole-4-carboxamide-1-beta-4-ribofuranoside in pancreatic cancer cells. Anticancer Res 30: 1875-1880. [Crossref]
- Penberthy DR, Rich TA, Shelton CH 3rd, Adams R, Minasi JS, et al. (2001) A pilot study of chronomodulated infusional 5-fluorouracil chemoradiation of pancreatic cancer. Ann Oncol 12: 681-4. [Crossref]
- Relles D, Sendecki J, Chipitsyna G, Hyslop T, Yeo CJ, et al. (2013) Circadian gene expression and clinicopathologic correlates in pancreatic cancer. J Gastrointest Surg 17: 443-450. [Crossref]
- Oda A, Katayose Y, Yabuuchi S, Yamamoto K, Mizuma M, et al. (2009) Clock gene mouse period2 overexpression inhibits growth of human pancreatic cancer cells and has synergistic effect with cisplatin. Anticancer Res 29: 1201-1209. [Crossref]
- Li XM (2010) Cancer inhibition through circadian reprogramming of tumour transcriptome with meal timing. Cancer Res 70: 3351-60. [Crossref]
- Balasenthil S, Chen N, Lott ST, Chen J, Carter J, et al. (2011) A migration signature and plasma biomarker panel for pancreatic adenocarcinoma. Cancer Res (Phila) 4:137-149. [Crossref]
- Brand RE, Nolen BM, Zeh HJ, Allen PJ, Eloubeidi MA, et al. (2011) Serum biomarker panels for the detection of pancreatic cancer. Clin Cancer Res 17: 805-816. [Crossref]
- Radon TP, Massat NJ, Jones R, Alrawashdeh W, Dumartin L, et al. (2015) Identification of a three-biomarker panel in urine for early detection of pancreatic adenocarcinoma. Clin Cancer Res 2: 3512. [Crossref]
- Wang J, Chen J, Chang P, LeBlanc A, Li D, et al. (2009) MicroRNAs in plasma of pancreatic ductal adenocarcinoma patients as novel blood-based biomarkers of disease. Cancer Prev Res (Phila) 2:807-813. [Crossref]
- Pedersen KS, Bamlet WR, Oberg AL, de Andrade M, Matsumoto ME, et al. (2011) Leukocyte DNA methylation signature differentiates pancreatic cancer patients from healthy controls. PLoS ONE 6: e18223. [Crossref]
- Wingren C, Sandström A, Segersvärd R, Carlsson A, Andersson R, et al. (2012) Identification of serum biomarker signatures associated with pancreatic cancer. Cancer Res 72: 2481-2490. [Crossref]
- Bracci PM, Zhou M, Young S, Wiemels J (2012) Serum autoantibodies to pancreatic cancer antigens as biomarkers of pancreatic cancer in a San Francisco Bay Area case-control study. Cancer 118: 5384-5394. [Crossref]
- Tanaka M, Javle M, Dong X, Eng C, Abbruzzese JL, et al. (2010) Gemcitabine metabolic and transporter gene polymorphisms are associated with drug toxicity and efficacy in patients with locally advanced pancreatic cancer. Cancer 116: 5325–5335. [Crossref]
- Woo HI, Kim KK, Choi H, Kim S, Jang KT, et al. (2012) Effect of genetic polymorphisms on therapeutic response and clinical outcomes in pancreatic cancer patients treated with gemcitabine. Pharmacogenomics 13: 1023–1035. [Crossref]
- Lemstrová R1, Souček P, Melichar B, Mohelnikova-Duchonova B (2014) Role of solute carrier transporters in pancreatic cancer: a review. Pharmacogenomics 15:1133-1145. [Crossref]
- Fujita H, Ohuchida K, Mizumoto K, Itaba S, Ito T, et al. (2010) Gene expression levels as predictive markers of outcome in pancreatic cancer after gemictabine-based chemotherapy. Neoplasia 12: 807-817. [Crossref]
- Tang SN, Fu J, Shankar S, Srivastava RK (2012) EGCG enhances the therapeutic potential of gemcitabine and CP690550 by inhibiting STAT3 signaling pathway in human pancreatic cancer. PLoS One 7: e31067. [Crossref]
- Li L, Leung PS (2014) Use of herbal medicines and natural products: An alternative approach to overcoming the apoptotic resistance of pancreatic cancer. Int J Biochem Cell Biol 53: 224–236. [Crossref]
- Boreddy SR, Srivastava SK2 (2013) Pancreatic cancer chemoprevention by phytochemicals. Cancer Lett 334: 86-94. [Crossref]
- Gong Z, Holly EA, Bracci PM (2009) Intake of folate, vitamins B6, B12 and methionine and risk of pancreatic cancer in a large population-based case-control study. Cancer Causes Control 20: 1317–1325. [Crossref]
- Donahue TR, Tran LM, Hill R, Li Y, Kovochich A, et al. (2012) Integrative survival-based molecular profiling of human pancreatic cancer. Clin Cancer Res 18: 1352-1363. [Crossref]
- Nakahara O, Takamori H, Tanaka H, Sakamoto Y, Ikuta Y, et al. (2010) Clinical significance of dihydropyrimidine dehydrogenase and thymidylate synthase expression in patients with pancreatic cancer. Int J Clin Oncol 15: 39–45. [Crossref]
- Neoptolemos JP (2009) Adjuvant 5-fluorouracil and folinic acid vs observation for pancreatic cancer: composite data from ESPAC-1 and -3(v1) trials. Br J Cancer 100: 246-250.
- Strippoli A (2016) ERCC1 expression effects outcome in metastatic pancreatic carcinoma treated with FOLFIRINOX: A single institution analysis. Oncotarget 7: 35159-35168. [Crossref]
- Li D, Liu H, Jiao L, Chang DZ, Beinart G, et al. (2006) Significant impact of homologous recombination DNA repair Gene polymorphisms on pancreatic cancer survival. Cancer Res 66: 3323–3330. [Crossref]
- Boeck S, Jung A, Laubender RP, Neumann J, Egg R, et al. (2013) EGFR pathway biomarkers in erlotinib-treated patients with advanced pancreatic cancer: translational results from the randomised, crossover phase 3 trial AIO-PK0104. Br J Cancer 108: 469-476. [Crossref]
- Vaz J, Ansari D, Sasor A, Andersson R (2015) SPARC: A Potential Prognostic and Therapeutic Target in Pancreatic Cancer. Pancreas 44: 1024-1035. [Crossref]