Background: Angelman syndrome (AS) is a neurodevelopmental disorder characterized by severe mental retardation, absent speech, dysmorphic facial features, microcephaly, epileptic seizures, Electroencephalography (EEG) abnormalities and neurological problem.
Four known molecular mechanisms lead to a deficiency in maternal UBE3A expression and consequently to AS: (1) Deletion of the AS critical region on the maternal chromosome 15q11.2–q13 (70%), (2) paternal uniparental disomy (pUPD) (2-7%), (3) imprinting defects (3–5%), and (4) mutations in the maternal copy of UBE3A (10%).
Materials and methods: Here, we report 11 Tunisian AS patients suspected on the basis of clinical features, behavior, EEG findings and confirmed by molecular analysis using FISH technique, microsatellites study and Methylation Specific Multiplex Ligation-dependent Probe Amplification (MS-MLPA).
Results: The diagnosis was confirmed in these patients (7 males, 4 females) by detecting the presence of deletion of the critical AS region on chromosome 15 through the use of fluorescence in situ hybridization (FISH) technique in 10 patients, and confirmed by Methylation-Specific Multiplex Ligation-dependent Probe Amplification (MS-MLPA). A microsatellite analysis detected only one patient with uniparental disomy.
Conclusion: Deletion and methylation aberration screening by MS-MLPA assay is considered as a rapid and cost-effective method to confirm Angelman syndrome diagnosis contributing to an early interventional therapy and genetic counseling should be provided.
Angelman syndrome, methylation disorders, 15q11-q13 locus, paternal uniparental
Abbreviations
AS: Angelman Syndrome; FISH: Fluorescence In-Situ Hybridization; UPD: Uniparental Disomy; MS-MLPA: Methylation-Specific Multiplex Ligation-dependent Probe Amplification; EEG: Electroencephalography; PWS: Prader-Willi syndrome; MRI: Magnetic Resonance Imaging; IC: Imprinting Center
Angelman syndrome (AS) (MIM #105830) is a neurogenetic disease affecting children, first identified in 1965 by a British doctor, Harry Angelman, from whom it also gained its name [1]. It is one of the most common genetic syndromes caused by non-mendelian inheritance in the form of genomic imprinting. The incidence of AS is estimated to be between 1/10,000 and 1/20,000, with equally affected males and females [2,3]. Cases have been reported from all over the world without racial predilection.
AS is attributable to the loss of gene expression within the chromosomal region 15q11-q13 [4]. The phenotype is well known in infancy and adulthood, but the clinical features may change with age. The main clinical characteristics include severe mental retardation, epileptic seizures and EEG abnormalities, neurological problems and distinct facial dysmorphic features. Behavioral problems such as hyperactivity and sleeping problems are reported, although these patients present mostly a happy personality with periods of inappropriate laughter [5]. The natural history is typified by normal growth parameters at the early age months. Developmental microcephaly appears by 2 years of age, delayed skill attainment by 6-12 months, speech impairment and seizures in 80% of cases by 3 years, and ataxic gait abnormalities [5]. Dysmorphic phenotypes are seen in about 80% of cases with occipital groove, flat occiput, protruding tongue, wide mouth, wide-spaced teeth, strabismus and prognathia [5]. Neuromuscular manifestations can include tongue thrusting, chewing/mouthing behaviors, swallow/suck disorders, truncal hypotonia during infancy, hyperactive lower extremity deep tendon reflexes, uplifted, flexed arm positioning during ambulation, and wide based gait with pronated or valgus-positioned ankles [5]. Abnormal sleep-wake cycles, heat intolerance, abnormal food related behaviors can also be seen [5]. The EEG is usually more abnormal than clinically expected, but it can also be normal in individuals with genetically proven AS [1].
Angelman syndrome is primarily due to maternal copy gene expression, lack of ubiquitin-protein ligase E3A (UBE3A) in fetal brain and adult frontal cortex. This can occur by maternal deletion of 15q11-13locus (68%), mutation in UBE3Agene (11%), paternal uniparental disomy (UPD) (7%), and imprinting center (IC) abnormalities (3%) [6]. However, 10% of AS patients have non diagnostic molecular findings [5].
There are a number of techniques that can confirm a diagnosis of AS. The detection of abnormal parent-specific methylation within the AS critical region has been reported in approximately 80% of individuals with AS [5]. The more commonly used methylation assessment techniques include Methylation Specific Multiplex Ligation-dependent Probe Amplification (MS-MLPA) and Southern blot analysis. The latter has greatly decreased out of favor due to the need for large amounts of high molecular weight DNA and the time and technical commitments required.MS-MLPA has the ability to determine aberrant methylation status and/or a deletion of the SNRPN locus. In the event of aberrant methylation but a normal diploid complement of the SNRPN locus, the haplotype studies are undertaken to discriminate between UPD and an imprinting defect especially of the imprinting center.
The purpose of this study was to evaluate the performance of MS-MLPA assay in a cohort of 11 Tunisian patients with definite diagnosis of AS, confirmed by Fluorescent in situ hybridization (FISH) or haplotype study; in order to assess concordance as well as the advantages and disadvantages of these laboratory techniques.
Patients
Eleven suspected patients by AS (7 males and 4 females) with average of 3.68), were referred to the Laboratory of Human Cytogenetic, Molecular Genetics and Reproductive Biology of Farhat Hached University Hospital, over a period of 11 years (2006 and 2017).
Patients were characterized with severe mental retardation, speech impairment, epileptic seizures or abnormal EEG findings and dysmorphic facial features (Table 1).
Table 1. Clinical and genetic features of 11 AS Tunisian patients
Case
n° |
Gender |
Age at diagnosis (year) |
Head Circumference |
Weight at diagnostic (Kg) |
Unsupported sitting |
Walking |
Speech
(single words) |
FISH/MS-MLPA results |
1 |
M |
7 |
47cm
(-4SD) |
23 |
3 Y |
4Y |
None |
15q11.q13 deletion |
2 |
M |
2.6 |
49 cm |
14 |
1Y |
instable |
None |
15q11.q13 deletion |
3 |
M |
5 |
47cm
(-2 SD) |
15 |
18 months |
instable |
None |
15q11.q13 deletion |
4 |
M |
1.8 |
47cm |
9 |
no |
no |
4.5 Y |
15q11.q13 deletion |
5 |
F |
6 |
46cm
(-2 SD) |
20 |
1Y 2 months |
5Y |
4.5 Y |
15q11.q13 deletion |
6 |
M |
6 |
50.5cm (+0.6 SD) |
23 |
4Y |
no |
2 Y |
15q11.q13 deletion |
7 |
F |
2.8 |
45cm
(-3.38 SD) |
12 |
no |
no |
None |
15q11.q13 deletion |
8 |
F |
3 |
46 cm
(-3 SD) |
13 |
6 months |
no |
None |
UPD |
9 |
M |
3 |
47.5cm
(-2 SD) |
15 |
7 months |
no |
None |
15q11.q13 deletion |
10 |
F |
3 |
47 cm |
14 |
1 Y |
3Y |
None |
15q11.q13 deletion |
11 |
M |
2 |
46 cm |
13.5 |
1Y |
no |
None |
15q11.q13 deletion |
M, male; F, female; Y: Year; SD: standard deviation; FISH: Fluorescence In Situ Hybridization; MS-MLPA: Methylation Specific-Multiplex Ligation -dependent Probe Amplification.
Consanguinity was documented in 4 families. patients belonging to 7 unrelated families were investigated as well and are originated from different regions of Tunisia. Consanguineous families were unrelated and originated from different regions of the country.
This study was approved by local ethics committees and performed with the patients and their families informed consent.
Conventional cytogenetic analysis
Chromosomal analysis was performed according to standard procedures for the patients and their parents. Briefly, peripheral blood lymphocytes were cultured in RPMI 1640 medium (Gibco®, Grand Island, NY, USA) enriched with 20% fetal calf serum, L-glutamine, antibiotics (penicillin and streptomycin) and phytohemagglutinin. The cells were cultured for 72 hours in a 37°C incubator with 5% CO2. Culture was stopped using colcemid solution (0.05 μg/mL) for 45 minutes. After harvesting, the cells were exposed to hypotonic solution (0.075 mol/L KCl) and fixed with methanol/acetic acid (3:1). The slides were prepared and stained using R-banding. A minimum of 20 metaphases were analyzed from each sample using the Applied imaging CytoVision Karyotyping System®. Karyotypes were assigned according to the recommendation of the International System of Human Cytogenetic Nomenclature (ISCN) 2005 [6,7].
Fluorescent in situ hybridization (FISH)
Analysis by fluorescence in situ hybridization (FISH) with probes D15Z1/SNRPN/PML [Vysis®, Downers Grove, IL] was performed according to supplier protocol. In all cases, we studied at least 10 metaphases and 50 interphases nuclei where we clearly identified hybridization to a control probes; D15Z1 at the centromeric region (spectrum green), and PML at 15q22-24 (spectrum orange) which can detect all possible translocations.
Ten microliters of probe were applied to metaphase slides and co-denaturized for 7 min at 75°C. After overnight hybridization at 37°C and washing, chromosomes were counterstained with 4,6 diamino-2-phenylindole (DAPI) and observed using an Axioskop Zeiss® fluorescentmicroscope and the images were captured with a CCD camera (Cytovision, AppliedImaging®).
We performed SALSA MLPA probemix kit P245-A2 (MRC-Holland, Amsterdam, Netherlands) that included probes targeting SNRPN and UBE3A genes, according to the method described and manufacturer’s recommendations [8]. This kit contains four probes located in the critical region of the PWS/AS and two control probes located at 15q24.1. Normalization was performed and the peak heights were compared to a synthetic control. Only when the peak was below 0.75 deletion was considered. For the analysis of fragments, we used ABI PRISM®310 Genetic Analyzer (Applied Biosystem), with ROX-500 as the internal size standard. Data interpretations were performed using Coffalyser (MRC-Holland) software.
Genotyping of polymorphic markers
PCR was carried out using the standard method with polymorphic markers located in the 15q11q13 region to verify paternal uniparental disomy. Eleven microsatellites markers were used (from centromere to telomere): D15S11, D15S646, D15S128, D15S1506, D15S122, D15S210, D15S986, D15S97, GABRB3, D15S966, D15S642 [9-10-11]. Eight of them (D15S11, D15S646, D15S128, D15S1506, D15S122, D15S210, D15S986 and GABRB3) were located within the critical region. PCR products were separated by capillary electrophoresis for 30 min on an ABI PRISM®310 Genetic Analyzer (Applied Biosystem) at 15 kV using a 36 cm capillary and POP4 polymer.
Clinical characteristics
Gender, age at diagnosis, main clinical features, cytogenetics and molecular analysis results of our patients are shown in table 1.
All patients showed severe speech deficit or absent speech, severe developmental delay, movement and behavioral abnormalities. Nine of them (81.81%) could sit without support, 2 (18.1%) could walk with support, and 9 could not walk. Eight of them (72.7%) had absence of speech, and 3 (27.2%) were able to speak a few meaningful words.
Dysmorphic facial features like occipital groove, protruding tongue, wide spaced teeth, prognathism were the most associated to our patients (Table 2). Ataxic gait was always present. All patients had seizures and were on antiepileptic therapy. Seizures were completely controlled in 3 patients, and partially in 8 (Table 2).
Table 2. Clinical features comparison between our 11 AS Tunisian patients and different AS patients with UPD/15q11q13 deletion reported in literature. Abbreviations: M: Male, F: Female, ND: Not Determined.
This study |
different studies |
Analyzed
parameters
at diagnosis |
Deletion
(10 patients) |
UPD
(1 patient) |
Values for 13 AS
patients
reported by
Tan et al [ 28] |
Values of one AS
patient with
UPD reported by
Horvath et al [29] |
Values of AS
patients with
deletion reported by
Varela Monica et al [27] |
Values of one
AS patient with
UPD reported by
Freeman et al [20] |
age (months) at diagnosis |
|
|
|
|
|
|
0-24 |
3 |
|
0 |
1 |
ND |
1 |
25-36 |
3 |
1 |
5 |
- |
ND |
- |
37-60 |
3 |
|
8 |
- |
ND |
- |
61-96 |
1 |
|
0 |
- |
ND |
- |
Gender |
7M/3F |
F |
8M/5F |
M |
29M/20F |
M |
Maternal age (Range) |
34 |
43 |
ND |
ND |
27 |
31 |
paternal age (years) |
38 |
49 |
ND |
ND |
32 |
ND |
birth weight (average-g) |
3135 |
2500 |
2140 |
3260 |
2981 |
4100 |
Hypotonia |
7/10 (70%) |
+ |
0/13 (0%) |
+ |
33/45 (73.33%) |
+ |
Neck support |
8/10 (80%) |
+ |
ND |
+ |
7 /12 |
+ |
sitting without support |
8/10 (80%) |
+ |
ND |
+ |
ND |
+ |
absent speech |
10/10 (100%) |
- |
13/13 (100%) |
+ |
43/47 (91.5%) |
+ |
Developmental delay |
10/10 (100%) |
+ |
12/13 (92%) |
+ |
49/49 (100%) |
+ |
Severe mental retardation |
10/10 (100%) |
+ |
12/13 (92%) |
+ |
49/49 (100%) |
+ |
Microcephaly |
8/10 (80%) |
+ |
8/13 (62%) |
+ |
25/46 (54.35%) |
- |
macrostomia |
7/10 (70%) |
- |
ND |
+ |
47/47 (100%) |
- |
Clinical seizures |
10/10 (100%) |
+ |
6/13 (46%) |
- |
42/47 (89.4%) |
- |
Occipital groove |
8/10 (80%) |
+ |
ND |
+ |
17/23 (73.9%) |
+ |
Protruding tongue |
8/10 (80%) |
+ |
ND |
+ |
28/40 (70%) |
- |
Wide-spaced teeth |
8/10 (80%) |
- |
9/13 (69%) |
+ |
34/39 (87.2%) |
+ |
Prognatism |
8/10 (80%) |
+ |
3/13 (23%) |
- |
ND |
+ |
unusually light hair or skin color |
3/10 (30%) |
+ |
3/13 (23%) |
- |
- |
+ |
Easily provoked laughter |
7/10 (70%) |
+ |
8/13 (62%) |
+ |
46/48 (95.8%) |
+ |
Hyperactivity |
8/10 (80%) |
+ |
ND |
- |
33/35 (94.3%) |
- |
Gastro-esophageal reflux |
5/10 (50%) |
- |
9/13 (69%) |
- |
ND |
- |
Ataxic gait |
10/10 (100%) |
+ |
8/13 (62%) |
- |
27/29 (93.1%) |
+ |
Frequent drooling |
5/10 (50%) |
+ |
10/13 (77%) |
+ |
32/33 (96.9%) |
+ |
History of sleep difficulties |
8/10 (80%) |
+ |
12/13 (92%) |
+ |
29/36 (80.5%) |
+ |
Fascination with water |
8/10 (80%) |
+ |
8/13 (62%) |
|
ND |
- |
Autistic behavior |
4/10 (40%) |
- |
0/13 (0%) |
- |
ND |
- |
Cranial MRI was performed in all the patients. Eight of them, were normal however 3 patients showed minimal cerebral atrophy (table 2). Metabolic screening tests revealed normal results in all patients.
Chromosomal analysis and FISH
Peripheral blood karyotypes of all the patients showed a normal chromosome complement: 46,XX or 46,XY according to gender (7 males and 4 females). Similarly, cytogenetic analysis of all the parents were normal, ruling out the presence of chromosomal rearrangement involving chromosome15.
FISH analysis in peripheral blood lymphocytes with specific probes (D15Z1/SNRPN/PML) revealed the presence of a deletion in the 15q11.2-q13 region in 10 patients (7 males and 3 females) (Table 1). The study of lymphocytes from their parents revealed a normal hybridization pattern and did not detect any deletion or rearrangement affecting chromosome, confirming the de novo 15q11.2-q13 deletion.
Microsatellite analysis
Only one patient no deleted-on FISH (patient N°8, Table 1) showed an inheritance of two paternal 15 chromosomes and no maternal chromosome 15 after the microsatellite analysis (Figure 1). The segregation analysis of the markers showed a homozygosity at all the locus on chromosome 15. He is classified as having paternal isodisomy.
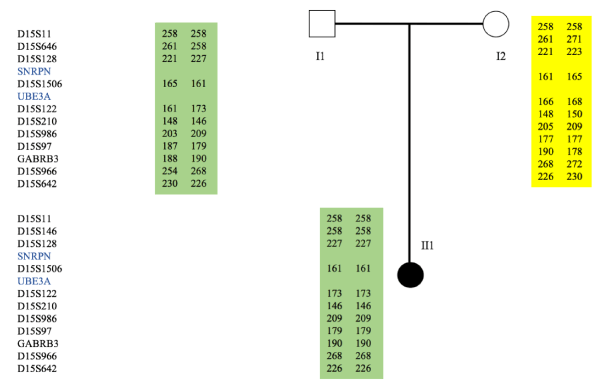
Figure 1. Microsatellites analysis and pedigree of the family with UPD patient. Microsatellites analysis of patient (II1) chromosome 15 fragments (D15S11, D15S646, D15S128, D15S1506, D15S122, D15S210, D15S986, D15S97, GABRB3, D15S966, D15S642, D15S11, D15S646, D15S128, D15S1506, D15S122, D15S210, D15S986) indicated that patient (II1) had inherited the same copies of 15 chromosomes from her father and no maternal chromosome 15
MS-MLPA analysis
In 10 patients, the maternal deletion 15q11.2-q13 was confirmed, while the dose for the control probes located on 15q24.1, was normal. The only patient with isoUPD, showed an aberrant methylation profile. The results of the parental blood DNAs were normal for all (data not shown).
Detection of complex epigenetic defects is a growing field in molecular diagnosis. There are a variety of approaches used in laboratories to confirm a clinical diagnosis of AS. These approaches include FISH, microsatellite analysis, Southern Blot and MS-MLPA, each with their own benefits and limitations. In this study, we performed a molecular analysis of MS-MLPA assay on 11 patients who had been referred for AS testing. The results obtained from the molecular techniques showed complete concordance, with an identical diagnosis in all the cases.
AS is a rare neurodevelopmental disorder with prevalence newly estimated between 1/22.000 and 1/52.000 [9-14], contrarily to previous studies which suggested prevalence about 1/10.000 to 1/20.000 [15-17]. The prevalence of AS in Tunisian population was not previously described in any research.
According to the study of Williams CA et al, the clinical features of AS are generally not important until late infancy [1]. Later, they are characterized by particular facial features with macrostomia and wide spaced teeth. Our group of patients showed normal birth weight and head circumference, normal deliveries at term. We found microcephaly in 80% of cases, severe mental retardation (100%), speech impairment (100%), ataxia (100%) and epilepsy (100%) (Table 2). All cases showed happy disposition with a constant smile or outbursts of laughter that sometimes look inappropriate, and an easily excitable personality with hand flapping, ataxic gait with arms flexed and upheld at the elbows, sleep disturbance, mirror reflection and love of water. The EEG was abnormal every time. Our results confirm those found by smith et al. [18].
Angelman syndrome is caused by epigenetic defects on 15q11-q13 Prader-Willi syndrome (PWS) and the AS critical region. It can be caused by different genetic mechanisms. Most of AS cases are due to 15q11q13 maternal deletion. The recurrence risk in this case is estimated to be <1% if mother chromosomes shows no rearrangements [19]. The second cause is the uniparental disomy of paternal chromosome 15. In remaining cases, UBE3A gene mutation or imprinting center abnormalities can be responsible of the disease. Determining the responsibility of one of them is crucial to provide accurate reproductive genetic counseling. Different techniques such as caryotype, FISH, Southern Blot, microsatellite analysis and MS-MLPA have been used in genetic diagnosis of AS.
In the present study, we analyzed eleven patients belonging to unrelated Tunisian families presented with a profile compatible with AS phenotype. Ten patients were confirmed by FISH to have a chromosome deletion of 15q11q13 and one patient have UPD confirmed by microsatellites haplotype study (table 1). He was homozygous at all loci for which his father was heterozygous, indicating that the structural rearrangement resulted from an isodisomic 15q. Few cases of AS resulting from isodisomic 15q associated UPD have been reported previously. The majority were heterodisomic. No phenotypic differences have been observed previously between the two mechanisms hetero or iso-UPD [20-23].
Multiplex ligation-dependent probe amplification (MLPA; MRC-Holland, Amsterdam, The Netherlands) is a technique for the detection of deletions and/or duplications in various genetic diseases [24,25]. For microdeletion syndromes, the MLPA kit allows the detection of copy number changes of several chromosomal regions (1p36, 7q11.23, 17p11.2, 17 p13.3, 22q11.21, 15q11.2, 20p12, 7p21.1, and 5q35.3). Thus, MLPA can significantly shorten the process by investigating various chromosomal regions at the same time. In this study, we introduced MLPA as a diagnostic test and then evaluated its performance compared to cytogenetic study and FISH.
The principle of MS-MLPA is almost similar to the MLPA, except that the target sequences detected by MS-MLPA probes contains a restriction site recognized by endonucleases such as HhaI or HpaII that are sensitive to cytosine methylation. Upon digestion with one of these enzymes, a probe amplification product will only be obtained if the CpG site is methylated. The level of methylation was determined by calculating the ratio of the relative peak area of each target probe from digested and undigested samples.
MS-MLPA results showed the heterozygote deletion for 10 patients previously detected by FISH. This deletion takes away the maternal functional copy of the 15q11q13 locus. The paternal remaining copy was methylated and non-functional. In one patient (N°8), we found an aberrant methylation profile.
Several aspects contribute to the benefit of MS-MLPA: (i) a large number of genes can be studied using a minimum amount of only 20 ng sample DNA, (ii) owing to its simple procedure, large number of samples can be analyzed simultaneously;(iii) MS-MLPA is semi-quantitative and can discriminate the copy number (deletion or duplication) and distinguish between methylation of one, both or none of the two alleles.
There is limited correlation between the clinical severity of AS and its type of genetic mechanism. Patients with the large chromosome deletions are more likely to have both microcephaly and seizures and are more likely to have eye, hair and skin hypopigmentation [26]. Those with uniparental disomy are more likely to have normal head circumference, no seizures, and better cognitive functioning, although severe to profound impairment is still present. Those with IC and UBE3A defects are more likely to have moderate clinical severity between the former and latter mechanisms stated above [1-27] described the clinical signs of 49 AS patients with deletion and concluded that the phenotypic variability observed in deletion patients can be observed in UPD patients but there are several differences between these two groups, include the gait improvement and the lower incidence of seizures in UPD. Haploinsufficiency of genes localized in the deleted chromosome segment is probably responsible for the more severe phenotypic and behavioral characteristics of deletion patients, as compared to patients with UPD, imprinting mutations and UBE3A gene mutations. These findings are in agreement with the observation that there is a less severe phenotype with UPD in our patient (Table 2). Although, our series of patients demonstrated a less severe phenotype, all showed severe speech delays and characteristic behaviors. No patients show more than a couple of meaningful words, and it can be that the core phenotype of AS is speech and/or expressive language dysfunction, along with characteristic behaviors.
By using MS-MLPA, the confirmation of the clinical diagnosis of AS can be promptly obtained. In addition, the copy number status can be determined concurrently with methylation status, thereby enabling cases with a deletion to be distinguished from those with UPD or an IC defect. Haplotype study may be required to determine if UPD is the cause of the syndrome or not.
MS-MLPA technique, used to detect the majority of aberrations abnormalities in AS, proves to be a robust, suitable and simple method for large-scale analysis of DNA to offer a correct genetic diagnosis indispensable for the comprehension of the disease mechanism and allowing a suitable genetic counseling.
We wish to express our appreciation for the cooperation and generosity of all contributing families. We would like to acknowledge the efforts of all team members in the Tunisian Department of Neuropediatry of La Rabta hospital. We also thank Mrs Ahlem Msakni, Mrs Sihem Sassi, and Mrs Safa Bouker for their excellent technical assistance.
- Williams CA (2003) Neurological aspects of the Angelman syndrome. Brain Dev 27: 88-94. [Crossref]
- Clayton-Smith J, Pembrey ME (1992) Angelman syndrome. J Med Genet 29: 412-415. [Crossref]
- Steffenburg S, Gillberg CL, Steffenburg U, Kyllerman M (1996) Autism in Angelman syndrome: a population-based study. Pediatr Neurol 14: 131-136. [Crossref]
- Buiting K, Williams C, Horsthemke B (2016) Angelman syndrome - insights into a rare neurogenetic disorder. Nat Rev Neurol 12: 584-593. [Crossref]
- Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, et al. et al. (2006) Angelman syndrome 2005: Updated consensus for diagnostic criteria. Am J Med Gen Part A 140: 413-418. [Crossref]
- Ramsden SC, Clayton-Smith J, Birch R, Buiting K (2010) Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet 11: 70. [Crossref]
- Shaffer LG, Slovak ML, Campbell LJ (2009) An international system for human cytogenetic nomenclature. Basel: Karger AG.
- Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, et al. (2002) Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 30: e57. [Crossref]
- Christian SL, Robinson WP, Huang B, Mutirangura A, Line MR, et al. (1995) Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am J Hum Genet 57: 40-48. [Crossref]
- Mutirangura A, Greenberg F, Butler MG, Malcolm S, Nicholls RD, et al. (1995) Multiplex PCR of three dinucleotide repeats in the Prader-Willi/Angelman criticalregion (15q11 q13): molecular diagnosis and mechanism of uniparental disomy. Hum Mol Genet 2: 143-151. [Crossref]
- Dib C, Fauré S, Fizames C, Samson D, Drouot N, Vignal A, et al. (1996) A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380: 152-154. [Crossref]
- Oiglane-Shlik E, Talvik T, Zordania R, Põder H, Kahre T, Raukas E, et al. (2006) Prevalence of Angelman syndrome and Prader-Willi syndrome in Estonian children: sister syndromes not equally represented. Am J Med Genet A 140: 1936-1943.
- Mertz LG, Christensen R, Vogel I, Hertz JM, Nielsen KB, et al. (2013) Angelman syndrome in Denmark. birth incidence, genetic findings, and age at diagnosis. Am J Med Genet Part A 161: 2197-2203. [Crossref]
- Luk HM, Lo, IF (2016) Angelman syndrome in Hong Kong chinese: A 20 years’ experience. Eur J Med Genet 59: 315-319. [Crossref]
- Kyllerman M (1995) On the prevalence of Angelman syndrome. Am J Med Genet 59: 405. [Crossref]
- Petersen MB, Brondum-Nielsen K, Hansen LK, Wulff K (1995) Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: Estimated prevalence rate in a Danish county. Am J Med Genet 60: 261-262. [Crossref]
- Buckley RH, Dinno N, Weber P (1998) Angelman syndrome: Are the estimates too low? Am J Med Genet 80: 385-390. [Crossref]
- Smith A, Marks R, Haan E, Dixon J, Trent RJ (1997) Clinical features in four patients with Angelman syndrome resulting from paternal uniparental disomy. J Med Genet 34: 426-429. [Crossref]
- Van Buggenhout GJ, Descheemaeker MJ, Thiry P, Trommelen JC, Hamel BC, et al. (2000) Angelman syndrome in three adult patients with atypical presentation and severe neurological complications. Genet Couns 11: 363-373. [Crossref]
- Freeman SB, May KM, Pettay D, Fernhoff PM, Hassold TJ (1993) Paternal uniparental disomy in a child with a balanced 15;15 translocation and Angelman syndrome. Am J Med Genet 45: 625-630. [Crossref]
- Robinson WP (2000) Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays 22: 452-459. [Crossref]
- Tonk V, Schultz RA, Christian SL, Kubota T, Ledbetter DH, et al. (1996) Robertsonian (15q;15q) translocation in a child with Angelman syndrome: evidence of uniparental disomy. Am J Med Genet 66: 426-428. [Crossref]
- Fridman C, Varela MC, Nicholls RD, Koiffmann CP (1998) Unusual clinical features in an Angelman syndrome patient with uniparental disomy due to a translocation 15q15q. Clin Genet 54: 303-308. [Crossref]
- Sellner LN, Taylor GR (2004) MLPA and MAPH: new techniques for detection of gene deletions. Hum Mutat 23: 413-419. [Crossref]
- Rusu C, Sireteanu A, Puiu M, Skrypnyk C, Tomescu E, et al. (2007) MLPA technique-principles and use in practice. Rev Med Chir Soc Med Nat Iasi 111: 1001-1004. [Crossref]
- Williams CA (2010) The behavioral phenotype of the Angelman syndrome. Am J Med Genet C Semin Med Genet 154: 432-437. [Crossref]
- Varela MC, Kok F, Otto PA, Koiffmann CP (2004) Phenotypic variability in Angelman syndrome: comparison among different deletion classes and between deletion and UPD subjects. Eur J Hum Genet 12: 987-992. [Crossref]