We report a 59-year old male patient who was referred from the Department of Urology complaining with recurrent flank pain for further imaging evaluation.
The patient had a known history of multiple cysts in both kidneys. However, previous imaging studies were not available at this time.
MR imaging of the abdomen was performed, showing multiple renal cysts on both sides. Some of the cysts also showed high protein or hemorrhagic shares. In addition, multiple liver parenchyma cysts and a diverticulosis coli were described (Figure 1).
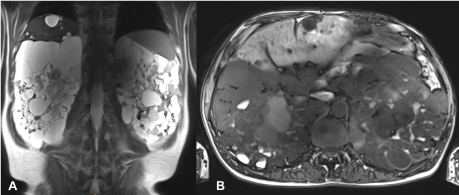
Figure 1. Initial MR examination: T2-weighted HASTE in coronal (a) and T1-weighted VIBE in transverse (b) orientation show multiple renal cysts with partial high protein or hemorrhagic content and multiple liver parenchyma cysts.
During the following three years, the patient was regularly checked in the urological ambulance. Due to recurrent but also increasing complaints and a marked restriction of lifestyle imaging follow-up studies were carried out in the meantime (Figure 2).
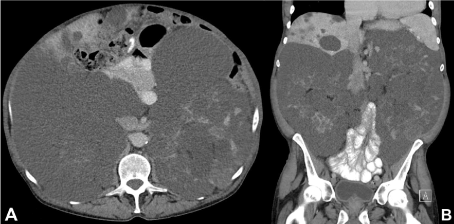
Figure 2. Follow-up contrast-enhanced (portal-venous phase) CT examination: Transverse (a) and coronal (b) images show the multiple renal and liver cysts with only slight progression.
In accordance with the previous MR scan, the non-contrast CT examinations (due to restricted renal function) showed clear signs of space-occupying cystic lesions with polycystic kidney, low-grade ascites and multiple liver cysts. Occasionally slightly enlarged lymph nodes were found.
In the further course, the patient eventually became dialysis dependent because of progressive renal failure. Afterwards the patient was listed for kidney transplant.
Due to the pronounced symptoms, a right sided nephrectomy was performed before the planned kidney transplantation.
The majority of polycystic kidney disease is caused by autosomal dominant or recessive inheritance, an acquired cystic kidney is rather rare (ACKD). ACKD usually develops in patients with chronic kidney failure and is associated with long-term dialysis [1]. The hereditary acquired polycystic kidney diseases include multiple disorders such as ADPKD, tuberous sclerosis, von-Hippel-Lindau syndrome, nephronophthisis, as well as some other very rare genetic disorders [2].
The most common form of polycystic kidney disease is the autosomal dominant polycystic kidney disease (ADPKD). It has an incidence of 1: 500 - 1: 1000 and is thus one of the most common hereditary diseases. It is caused by a mutation of the PKD-1 gene (85%) or the PKD-2 gene (15%) [3].
The diagnosis is a clinical one. The following factors are decisive: age, positive family history, enlarged kidneys with multiple cysts on both sides and a slightly limited renal function at the time of diagnosis. Usually, onset of the disease is between 30 - 50 years. The detection of liver cysts and other extrarenal manifestations may help in the diagnosis with missing or negative family history [2].
The most common extrarenal manifestation of ADPKD are multiple liver parenchyma cysts [4]. Furthermore, a diverticulosis coli is a common feature [5].
In addition, pancreatic cysts, heart valve changes and aneurysms of the aorta plus cerebral arteries can be found [2].
The most common renal complications include arterial hypertension, cyst infections and refractory pain. The flank pain is usually caused by the increasing intraabdominal pressure, but also by cyst rupture and infections [6].
Even today, ADPKD can only be cured with kidney transplant. Only one drug is currently approved for treatment of ADPKD, a V2-vasopression receptor antagonist (Tolvaptan) [7].
No conflicts of interest. No grants or financial supports.
- Matson MA, Cohen EP (1990) Acquired cystic kidney disease: occurrence, prevalence, and renal cancers. Medicine (Baltimore) 69: 217-226. [Crossref]
- Kahn W, Walz G. (2007) Autosomal dominante polyzystische Nierenerkrankung. Dtsch Arztebl Int. 104(44): A-3022-3028.
- Wilson PD (2004) Polycystic kidney disease. N Engl J Med 350: 151-164. [Crossref]
- Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, et al. (1990) Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology 11: 1033-1037. [Crossref]
- Sharp CK, Zeligman BE, Johnson AM, Duley I, Gabow PA (1999) Evaluation of colonic diverticular disease in autosomal dominant polycystic kidney disease without end-stage renal disease. Am J Kidney Dis 34: 863-868. [Crossref]
- Bajwa ZH, Sial KA, Malik AB, Steinman TI (2004) Pain patterns in patients with polycystic kidney disease. Kidney Int 66: 1561-1569. [Crossref]
2021 Copyright OAT. All rights reserv
- Gansevoort RT, Arici M, Benzing T, Birn H, Capasso G, Covic A, et al. (2016) Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice. Nephrol Dial Transplant 31(3): 337-348. [Crossref]