Chronic myeloid leukemia (CML) is effectively treated with tyrosine kinase inhibitors (TKI) targeted against BCR-ABL. The second-generation of ABL-tyrosine kinase inhibitors (2nd TKIs) can be more promising approach for efficient reduction of the CML stem cells. However, there are cases of relapse and refractoriness despite the use of existing TKIs, and cases of intolerance despite good response to therapy, in which the emergence of mutations during TKI treatment is one of the factors, especially T315I mutation, which is resistant to up to the 2nd TKIs. Although ponatinib, a third-generation TKI, is effective in patients refractory to the 2nd TKIs, including those expressing T315I, ponatinib is associated with increased cardiovascular side effects compared with imatinib. Moreover, we need to develop the evaluation method of the residual CML diseases and novel therapies to establish rational therapy-cessation strategies in CML. In this review, we focus the novel generation of molecular target therapies for CML: STAMP (Specifically Targeting the ABL Myristoyl Pocket) inhibitor Asciminib and medium-chain fatty-acid derivative AIC-47.
BCR-ABL, chronic myeloid leukemia, leukemia stem cells, tyrosine kinase inhibitors, asciminib
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder that is characterized by the presence of a fusion oncogene, BCR-ABL, which encodes a protein with constitutive tyrosine kinase activity [1]. CML is effectively treated with tyrosine kinase inhibitors (TKI) targeted against BCR-ABL such as imatinib (IM). In the nonrandomized Stop Imatinib (STIM) study, IM treatment was discontinued in patients with CML who had achieved complete molecular remission (CMR) of more than 2-year duration [2]. Of the 69% of patients with complete follow-up, 61% relapsed from CMR states (nevertheless, all patients who relapsed responded safely to the reintroduction of IM). The remaining patients maintained CMR states, suggesting that TKI treatment may cure some proportion of patients with CML [3,4]. Ross, et al. proposed the sensitive measurement of minimal residual disease using genomic PCR method with patient-specific primers [5]. We previously reported the investigation of residual CML diseases during TKI treatment using FACS-sorting and quantitative RT-PCR of BCR-ABL among each population; total mononuclear cells, hematopoietic stem cells, and myeloid progenitors [6]. The observations also implied that the second-generation of ABL-tyrosine kinase inhibitors (2nd TKIs), dasatinib or nilotinib therapy can be more promising approach for efficient reduction of the CML stem cells. However, there are cases of relapse and refractoriness despite the use of existing TKIs, and cases of intolerance despite good response to therapy, in which the emergence of mutations during TKI treatment is one of the factors, especially T315I mutation, which is resistant to up to second generation TKIs. Although ponatinib, a third-generation TKI, is effective in patients refractory to second-generation TKIs, including those expressing T315I, ponatinib is associated with increased cardiovascular side effects compared with imatinib [7-9], and intolerance associated with these side effects makes it difficult for some patients to continue treatment. Moreover, we need to develop the evaluation method of the residual CML diseases to establish rational therapy-cessation strategies in CML.
In this review, we focus the novel generation of molecular target therapies for chronic myeloid leukemia: STAMP (Specifically Targeting the ABL Myristoyl Pocket) inhibitor Asciminib and medium-chain fatty-acid derivative AIC-47.
Both of first and second generation TKIs have improved prognosis of patients with CML. However CML cells acquire resistance during these TKI treatments. The resistance or intolerance mainly occurs because of BCR-ABL mutations following replacements of amino acids in BCR-ABL. Replacements of amino acids are observed in ABL tyrosine kinase domains. The structure of BCR-ABL changed and these induce the lack of TKIs binding to ABL. Classic TKIs target the BCR-ABL ATP binding catalystic site and reduce tyrosine kinase activities. Several mutations were reported [10], and second generation TKIs against most of mutations except for T315I mutation which located in the middle of ATP binding site. Only ponatinib overcomes T315I mutation. However new options are needed for patients with resistance or tolerance to ATP binding TKIs or who do not achieve treatment goals with them.
The small molecules were reported to bind to the BCR-ABL myristoyl pocket and inhibit tyrosine kinase activity via an allosteric mechanism [11] (Figure 1). Unfortunately the first small molecule had limited potency and lacked activity against the BCR-ABL T315I mutation. However second generation allosteric inhibitors have similar cellular potencies but distinct patterns of resistance mutations [12,13]. Asciminib (ABL001) is a potent and specific inhibitor of BCR-ABL with a mutation profile complementary to that of ATP-binding TKIs. Asciminib and nilotinib which is a second generation ATP-binding TKI did not share the resistance with genetic barcoding studies [13]. In this study, rapid tumor regression was seen with single-agent asciminib or nilotinib in the mice xenograft model. However, the resistant emerged due to the A337V and P223S mutations with asciminib and T315I mutations with nilotinib. On the other hand, sustained tumor regression was observed with combination treatment of asciminib and nilotinib. Based on this study, clinical trials performed to explore asciminib in patients with CML-CP.
Figure 1. The pharmacokinetics of the allosteric ABL1 inhibitor
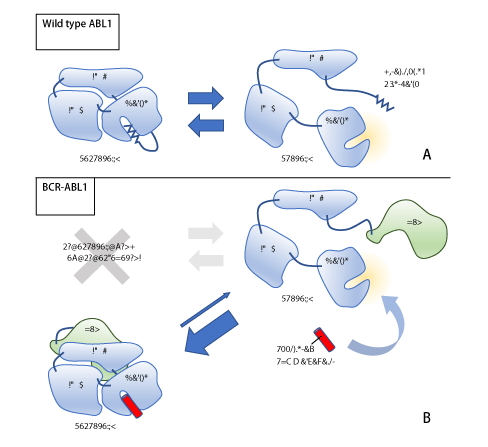
The Myristoylated N-termina of wild type ABL1 binds to the active kinase domain, which transform ABL1 to the inactive form (Panel A). The fusion protein BCR-ABL1 constantly stays in the active form because of the absence of the inhibitory N-terminal. The allosteric ABL1 inhibitor binds to the kinase domain mimicking the myristoylated portion and results in inactivation of BCR-ABL1
In phase 1 open-label study (NCT02081378) to determine the maximum tolerated dose or the recommended dose of asciminib, 141 patients with chronic-phase and 9 with accelerated-phase chronic myeloid leukemia (CML) who had resistance to or unacceptable side effects from at least two previous ATP-competitive tyrosine kinase inhibitors (TKIs) were enrolled. The median follow-up was 14 months [14]. Although 70% (105 of 150 patients) had received at least three TKIs, among patients with CML-CP, 34 (92%) with a hematologic relapse had a complete hematologic response; 31 (54%) without a complete cytogenetic response at baseline had a complete cytogenetic response. A major molecular response was achieved or maintained by 12 months in 48% of patients who could be evaluated, including 8 of 14 (57%) deemed to have resistance to or unacceptable side effects from ponatinib. A major molecular response was achieved or maintained by 12 months in 5 patients (28%) with a T315I mutation at baseline. The maximum tolerated dose of asciminib was not reached. Dose-limiting toxic effects included asymptomatic elevations in the lipase level and clinical pancreatitis. Common adverse events included fatigue, headache, arthralgia, hypertension, and thrombocytopenia, of which 92% were grade 1 or 2. This study showed Asciminib was active in heavily pretreated patients with CML who had resistance to or unacceptable side effects from TKIs, including patients in whom ponatinib had failed and those with a T315I mutation.
Following that previous results, phase 3 trial (NCT03106779) to verify whether asciminib could provide superior efficacy to bostinib beyond 2nd line was conducted [15].
In this phase 3, open-label study, patients with CML-CP previously treated with ≥2 TKIs were randomized (2:1) to receive asciminib 40 mg twice daily vs bosutinib 500 mg once daily. Randomization was stratified by major cytogenetic response (MCyR) status at baseline. The primary endpoint was major molecular response (MMR) rate at 24 weeks with asciminib vs bosutinib. Two hundred thirty-three patients were randomized to receive asciminib 40 mg twice daily (n=157) or bosutinib 500 mg once daily (n=76). Median follow-up was 14.9 months. MMR rate at 24 weeks was 25.5% with asciminib vs 13.2% with bosutinib. The between-arm common treatment difference, after adjusting for MCyR at baseline, was 12.2% (95% CI, 2.19-22.3), which was statistically significant; 2-sided P=0.029. Grade ≥3 adverse events occurred in 50.6% and 60.5% of patients receiving asciminib and bosutinib, respectively.
TKIs have greatly improved the prognosis for patients with CML; however, some patients experience treatment failure. In particular, the T315I mutant is uniformly resistant to TKIs other than ponatinib [16]. A recent study showed that long-term outcome of patients with ponatinib failure are poor with 1-year overall survival and 1-year event-free survival [17], and new treatment options are needed for these patients.
Medium-chain fatty-acid derivative AIC-47 is a novel anti-cancer agent for the CML therapy [18-21] (Figure 2). TKIs inhibit only the phosphorylation of downstream signaling pathway of BCR-ABL, whereas, AIC-47 decreases the expression of BCR-ABL itself, which is the greatest difference between AIC-47 and TKIs. One of the mechanisms of the decreased expression of BCR-ABL by AIC-47 is transcriptional repression of BCR-ABL gene [18]. As the effects of AIC-47 are independent of the structure of BCR-ABL tyrosine kinase domain, AIC-47 has anti-leukemic effects in either wild-type- or mutated-BCR-ABL-harboring cells in vitro and in vivo [20]. We recently found that BCR-ABL protein is degraded by autophagy, and T315I-BCR-ABL is insusceptible to autophagic degradation [21] (Figure 3). The increased stability of T315I-BCR-ABL might be one of the mechanisms of resistance to TKIs. We found that AIC-47 is an autophagy inducer, and that it suppresses BCR-ABL expression via autophagic degradation [21].
Figure 2. Discovery of medium-chain fatty-acid derivatives as novel anti-cancer agents
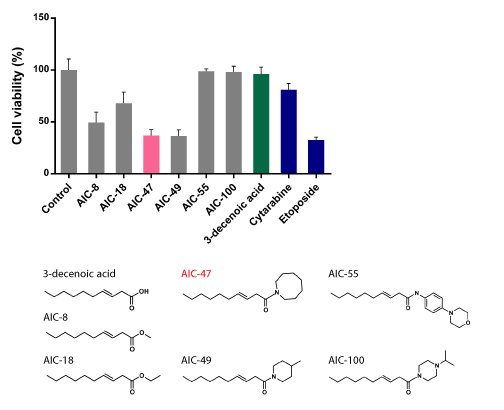
Cytotoxic effect of medium-chain fatty-acid derivatives in human CML K562 cells. More than 800 compounds were tested and several compounds, of which structure is shown in the lower panel, have anti-cancer effects. K562 cells were treated with 5 μM of each compound and viable cells were evaluated. The cell viability of the Control (DMSO alone) is indicated as 100%. The activities of modified compounds including AIC-47 are more enhanced compared to scaffold structure 3-decenoic acid (green bar), and are approximately the same as the activity of Etoposide (blue bar)
Figure 3. AIC-47 mode of action
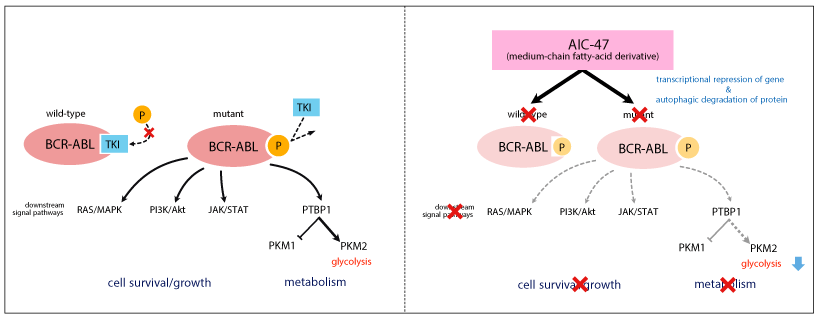
TKIs traps the BCR-ABL kinase in an inactivate conformation. The point mutations including T315I inhibits the interface between BCR-ABL and TKIs (left panel). Medium-chain fatty-acid derivative AIC-47 is effective for CML cells with both wild-type and mutated-BCR-ABL. AIC-47 induces transcriptional repression of BCR-ABL gene and promotes autophagic degradation of BCR-ABL protein. The downstream signals are inhibited and cancer-specific energy metabolism is disrupted by AIC-47, whose effects are independent of the configuration of BCR-ABL kinase (right panel)
Anti-cancer drugs targeting the energy metabolism of cancer is recently expected to be a new strategy for overcoming drug resistance. One of the hallmarks of cancer glycolytic metabolism “Warburg effect” is achieved through regulated expression of pyruvate kinase isoforms, PKM1 and PKM2, by alternative splicers including PTBP1 [22,23] (Figure 4). We found that knockdown of BCR-ABL leads to perturbation of the Warburg effect through the PTBP1/PKM cascade [18]. Glucose metabolism is a central source of energy for cancer, and BCR‐ABL activates glycolysis and promotes glucose‐dependent survival [24]. These findings indicate that BCR-ABL functions as one of the key molecules of glycolysis in CML. The PTBP1/PKM cascade is conserved even in BCR-ABL-mutated TKI resistant cells [20], suggesting that the cascade could be a potential target for overcoming TKI resistance or intolerance in CML. As a consequence of the down-regulation of BCR-ABL, AIC-47 changes the ratio of PKM1/PKM2 through the down-regulation of PTBP1, and perturbs the glycometabolism in CML cells regardless of the mutation of BCR-ABL [20].
Figure 4. A hallmark of cancer glycolytic metabolism “Warburg effect” in CML
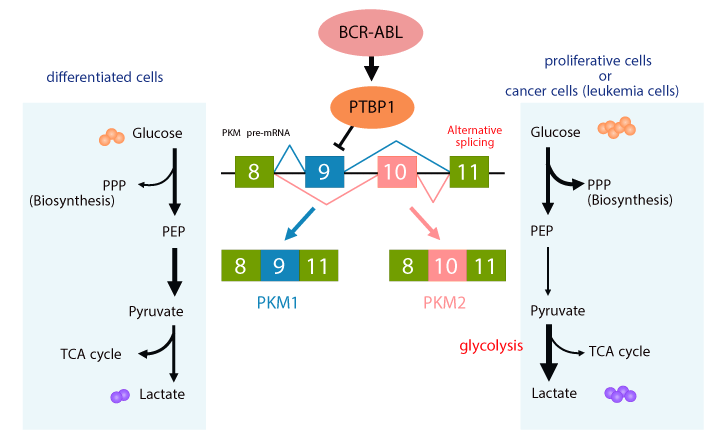
Proliferative cells and cancer cells including CML consume glucose more avidly, and convert glucose to lactate by glycolysis even in an aerobic environment, which is the well-known “Warburg effect.” The Warburg effect is partly achieved through control of pyruvate kinase isoforms PKM1 and PKM2 expression. These two isoforms result from alternative splicing of the PKM pre-mRNA, reflecting the inclusion of either exon 9 (PKM1) or exon 10 (PKM2). PTBP1 excludes exon 9, resulting in exon 10 inclusion and PKM2 expression. PKM2 converts phosphoenolpyruvate (PEP) to pyruvate less efficiently than PKM1, which contributes to biosynthesis for cell proliferation through the pentose phosphate pathway (PPP). BCR-ABL positively regulates PTBP1 and PKM2 expression, and it is conserved even in BCR-ABL-mutated cells
The preparation of this review was partially supported by the National Cancer Research Grants & the Grants-in-Aid from the National Institute of Biomedical Innovation and from the Ministry of Education, Culture, Sports, Science and Technology on Scientific Research, Japan.
Y Minami received research funding from Ono and CMIC, and honoraria from Bristol-Myers Squibb, Novartis, Astellas, and Daiichi-Sankyo; T Ono received research funding from Celgene, Chugai Pharmaceutical Co.,Ltd. and Kyowa Hakko Kirin, and honoraria from Bristol-Myers Squibb, Novartis, Pfizer and Otsuka Pharmaceutical Co., Ltd.; the other authors declare no conflicts of interest.
- Melo JV, Barnes DJ (2007) Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer 7: 441-453. [Crossref]
- Mahon F-X, Réa D, Guilhot J, Guilhot F, Huguet F, et al. (2010) Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 11: 1029-1035. [Crossref]
- Deininger M (2011) Hematology: curing CML with imatinib--a dream come true? Nat Rev Clin Oncol 8: 127-128. [Crossref]
- Pellicano F, Sinclair A, Holyoake TL (2011) In search of CML stem cells' deadly weakness. Curr Hematol Malig Rep 6: 82-87.
- Ross DM, Hughes TP, Melo JV (2010) Do we have to kill the last CML cell? Leukemia 25: 193-200.
- Minami Y, Abe A, Minami M, Kitamura K, Hiraga J, et al. (2012) Retention of CD34+ CML stem/progenitor cells during imatinib treatment and rapid decline after treatment with second-generation BCR-ABL inhibitors. Leukemia 26: 2142-2143. [Crossref]
- Moslehi JJ, Deininger M (2015) Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J Clin Oncol 33: 4210-4218. [Crossref]
- Douxfils J, Haguet H, Mullier F, Chatelain C, Graux C, et al. (2016) Association Between BCR-ABL Tyrosine Kinase Inhibitors for Chronic Myeloid Leukemia and Cardiovascular Events, Major Molecular Response, and Overall Survival: A Systematic Review and Meta-analysis. JAMA Oncol 2: 625-632. [Crossref]
- Valent P, Hadzijusufovic E, Schernthaner GH, Wolf D, Rea D, et al. (2015) Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood 125: 901-906. [Crossref]
- Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, et al. (2011) BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood 118: 1208-1215. [Crossref]
- Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, et al. (2006) Allosteric inhibitors of Bcr-abl–dependent cell proliferation. Nat Chem Biol 2: 95-102. [Crossref]
- Zhang J, Adrián FJ, Jahnke W, Cowan-Jacob SW, Li AG, et al. (2010) Targeting Bcr–Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463: 501-506. [Crossref]
- Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, et al. (2017) The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nature 543: 733-737. [Crossref]
- Hughes TP, Mauro MJ, Cortes JE, Minami H, Rea D, et al. (2019) Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N Engl J Med 381: 2315-2326.
- Andreas Hochhaus M, Carla Boquimpani M, Delphine Rea M, Minami Y, Lomaia E, et al. (2020) Efficacy and Safety Results from ASCEMBL, a Multicenter, Open-Label, Phase 3 Study of Asciminib, a First-in-Class STAMP Inhibitor, vs Bosutinib (BOS) in Patients (Pts) with Chronic Myeloid Leukemia in Chronic Phase (CML-CP) Previously Treated with ≥2 Tyrosine Kinase Inhibitors (TKIs. Blood Supplement 2: LBA-4.
- O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, et al. (2009) AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16: 401-412. [Crossref]
- Boddu P, Shah AR, Borthakur G, Verstovsek S, Garcia-Manero G, et al. (2018) Life after ponatinib failure: outcomes of chronic and accelerated phase CML patients who discontinued ponatinib in the salvage setting. Leuk Lymphoma 59: 1312-1322. [Crossref]
- Shinohara H, Taniguchi K, Kumazaki M, Yamada N, Ito Y, et al. (2015) Anti-cancer fatty-acid derivative induces autophagic cell death through modulation of PKM isoform expression profile mediated by bcr-abl in chronic myeloid leukemia. Cancer Lett 360: 28-38. [Crossref]
- Shinohara H, Kumazaki M, Minami Y, Ito Y, Sugito N, et al. (2016) Perturbation of energy metabolism by fatty-acid derivative AIC-47 and imatinib in BCR-ABL-harboring leukemic cells. Cancer Lett 371: 1-11. [Crossref]
- Shinohara H, Sugito N, Kuranaga Y, Heishima K, Minami Y, et al. (2019) Potent antiproliferative effect of fatty-acid derivative AIC-47 on leukemic mice harboring BCR-ABL mutation. Cancer Sci 110: 751-760. [Crossref]
- Shinohara H, Minami Y, Naoe T, Akao Y (2019) Autophagic degradation determines the fate of T315I-mutated BCR-ABL protein. Haematologica 104: e191-e194. [Crossref]
- Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC (2008) Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452: 181-186. [Crossref]
- Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, et al. (2010) The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci U S A 107: 1894-1899. [Crossref]
- Barger JF, Gallo CA, Tandon P, Liu H, Sullivan A, et al. (2012) S6K1 determines the metabolic requirements for BCR-ABL survival. Oncogene 32: 453-461. [Crossref]