Abstract
Systemic Juvenile Idiopathic Arthritis (SJIA) or Still's disease consists of an inflammatory systemic process without known etiology. It affects both genders in the same proportions, and represents 8.9% of Juvenile Idiopathic Arthritis's cases. The clinical findings vary, although fever of unknown origin is common. This study describes a 6 years old boy diagnosed with SJIA, characterizing the manifestations of this rheumatologic disease, the main signs and symptoms, laboratory evaluation, diagnostic criteria and treatment.
Key words
still's disease, systemic juvenile idiopathic arthritis, fever of unknown origin, case report
Introduction
Systemic Juvenile Idiopathic Arthritis (SJIA) is one of the leading causes of short- and long-term disability, affecting approximately 250,000 children in the United States [1]. This disease can be classified according to the clinical presentation, which is categorized into seven subtypes by the International League of Associations for Rheumatology (ILAR): A) oligoarticular; B) positive rheumatoid factor (RF) polyarticular; C) negative polyarticular RF; D) arthritis associated with enthesitis; E) juvenile psoriatic arthritis; F) undifferentiated arthritis and G) Systemic JIA [2].
The systemic variant of SJIA, also known as Still's Disease, is characterized as a multisystemic inflammatory disease, with daily fever, cutaneous rash, arthritis, pericarditis, hepatosplenomegaly, lymphadenopathy, arthralgia and intense myalgia [3]. The laboratory tests showed leukocytosis (greater than 15,000 cells/mm3), normocytic and normochromic anemia, changes in the level of serum liver enzymes, increase of acute phase inflammatory proteins, resulting in elevated levels of serum ferritin [4]. Its etiology, according to Pascual et al. [1], remains unclear, but the literature demonstrate that there is an increase in IL-6 levels, which may be related to the systemic activation of the disease. The differential diagnosis is made excluding infectious diseases, neoplasms and collagenosis.
This article aims to report the case of a school patient with the diagnosis of Still's Disease, characterizing the manifestations of this rheumatologic disease, the main signs and symptoms, laboratory evaluation, diagnostic criteria and treatment.
Case report
CSMP, 6-year-old boy from São Paulo, resident of Maringá, reported a previous history of intermittent high fever for 10 days (1 to 2 daily episodes of 39°C to 40°C) and diffuse, moderate to severe intensity, abdominal pain. It was initiated oral antimicrobial treatment (amoxicillin - 50 mg/kg/day) and then the patient developed cutaneous rash (Figure 1) with persistence of symptoms. Opted for exchange the antibiotic scheme to azithromycin (500 mg/kg/day for 3 days) and associated ibuprofen (200 mg/dose at 6/6 h) by oral route. During this period, despite the medication, the patient presented a new skin rash and worse of abdominal pain. He was referred to our hospital using ceftriaxone (80 mg/kg/day) empirically and suspected monolike syndrome.
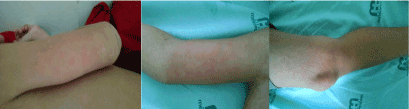
Figure 1: Cutaneous rash manifestations
At physical examinatio2021 Copyright OAT. All rights reservbrile, eupneic and pale 2+/4. The abdomen was tense, the airway sounds diminished, painful to the superficial and deep palpation (mainly on flanks).
On admission, the following laboratory tests were requested: blood count, C-reactive protein, renal and hepatic function, and amylase. Complementary exams include microcytic and hypochromic anemia: 7.8 mg/dL, leucocytosis: 20,300/mm3 and plaques: 521,000/mm3, C-reactive protein: 80.18 mg/dL. The cultures (blood culture, uroculture and coproculture) and cytomegalovirus, toxoplasmosis and Epstein Barr Virus serologies were negative, excluding the hypothesis of infectious diseases. Image exams such as computed tomography of the abdomen with and without contrast, abdominal radiography and transthoracic echocardiogram showed no alterations. Abdominal ultrasound revealed signs suggestive of inflammatory bowel disease and echoDoppler showed signs of mesenteric adenitis with more than 10 enlarged lymph nodes. Fecal occult blood testing was negative. ANA (anti-nuclear antibody) and rheumatoid factor test (RF), ferritin and LDH (lactic dehydrogenase) were investigated. Ferritin and LDH were significantly altered: 2,161 ng/ml and 668 IU/l, respectively. ANA and RF presented negative results.
During hospitalization, the patient remained febrile, with nocturnal peaks of 38ºC to 39ºC, evanescent rash, diffuse abdominal pain and ankle arthralgia. With the persistence of symptoms, medications such as metronidazole (20 mg/dose 6/6 h for 7 days) and albendazole (400 mg/dose/day for 3 days) were started, but with no improvement. Due to the presence of persistent fever of indeterminate origin, evanescent rash, abdominal pain and the exclusion of the infectious or neoplastic etiology, the diagnosis of Still's disease was suspected.
Thus, on the 11th day of hospitalization, non-steroidal anti-inflammatory oral therapy (indomethacin - 50 mg/day) was initiated. After the beginning of the treatment, the patient presented only a peak of fever, remaining afebrile ever since and improving abdominal pain. The onco-pediatrician evaluation was requested to proceed with a myelogram which, however, was unchanged. After the rheumatologist's evaluation, a conclusive diagnosis of Still's syndrome was made, and corticoid (prednisolone – 4 mg/kg/day)) and methotrexate (20 mg/m2 twice a week) oral therapy were initiated.
Discussion
Juvenile idiopathic arthritis is a rare rheumatologic disease of unknown etiopathogenesis [5,6]. According to the American College of Rheumatology, it can present as: systemic, pauciarticular or polyarticular, according to the main clinical manifestations in the first six months of the disease [7]. Its clinical course is variable and may remit in almost 50% of the cases, and between the three types, the systemic form is probably the one that most drive us to an extensive investigation of differential diagnosis [2,6]. This has no predilection by gender and is the least common onset form, also known as Still's disease. It mainly affects children under 4 years of age, prone to protein and caloric malnutrition secondary to anorexia, with low nutrient intake and increased catabolism of lipids and proteins [7]. Often, their initial manifestations are extra-articular and may overlap or precede Arthritis, which emphasizes its systemic nature.
It is usually characterized by high fever, evanescent rash and arthritis, in addition to the multisystemic involvement: generalized lymphadenopathy, hepatosplenomegaly, altered liver function tests, neutrophilic leukocytosis, normocytic anemia, increased CRP, pneumonitis, pleural effusion and pericarditis. Renal and neurological involvement are rare. Serum ferritin levels above 1000 ng/ml correlate with disease activity [5]. The presence of rheumatoid factor is not usual, and this is not used to diagnosis or prognosis. The ANA is rarely detectable, complements (C3, C4 and CH50) are normal and have no relation to genetic markers [6]. The most involved joints are the knee, wrist and ankle. However, arthritis may occur in small joints of the hands and in the axial skeleton [2]. Although arthritis is the cornerstone to perform the diagnosis of JIA, it is known that, in its systemic form, joint manifestations may not be concomitant with the appearance of the clinical picture.
In their study, Rosmaninho et al. [6] report that in 1/3 of the cases, patients only present with arthralgia and the arthritis is only checked a little later. There is a great variety in the severity of the symptoms, from fever and rash for two to three weeks followed by discrete arthritis, until the simultaneous appearance of all the aforementioned symptoms. The lack of specific markers requires differential diagnoses with infections, collagenoses, inflammatory and neoplastic diseases [2]. However, even though it is a diagnosis of exclusion, its diverse range of laboratory alterations made it possible to formulate diagnostic criteria from the description of the disease in 1971 by Bywaters [8].
The established treatment for systemic JIA is initiated with non-steroidal anti-inflammatory drugs (NSAIDs) and systemic corticosteroids. Methotrexate is an alternative in the treatment [2]. Indomethacin or acetylsalicylic acid may be used. If there is no response to the use of NSAIDs, the use of corticosteroids is started, and good clinical and laboratory evolution are described in the literature. For patients who do not ameliorate to conventional therapy, other agents, such as hydroxychloroquine, gold salts and methotrexate (MTX) are used, the latter having beneficial effects in the systemic form of the disease. The use of pulse therapy with methylprednisolone is reserved for particular cases [5,6]. The period of treatment of Still's disease is somewhat unclear in the literature because, since the disease has an alterable character according to each patient, establishing a fixed time for therapy is not feasible. Some reports discuss the use of NSAIDs for 4 to 6 months ago, while others limit treatment to remission of symptoms.
It is known, however, that the disease can evolve in a variety of ways, ranging from total remission of symptoms to hepatic failure, amyloidosis, cardiac failure, status epilepticus, disseminated intravascular coagulation and thrombotic thrombocytopenic purpura [2]. Delayed growth may be related not only the disease itself, but also the prolonged use of corticosteroids [6]. However, the most urgent complication of Still disease is macrophagic activation syndrome (MAS), also present in several other rheumatologic diseases such as systemic lupus erythematosus and ankylosing spondylitis, its prevalence in systemic JIA is approximately 7% [2]. MAS associated with JIA was initially described by Hadchouel et al. [9].
The clinical is based on prolonged high fever, hepatosplenomegaly, bleeding, generalized adenomegaly, exanthems, jaundice, and may develop with acute hepatic failure, encephalopathy, coma, disseminated intravascular coagulation (DIC) and multiple organ failure [10]. The presence of numerous macrophages in the bone marrow, phagocytizing blood cells (hemophagocytosis), without evidence of malignancy is its hallmark. Initially, the treatment is based on discontinuation of all non-hormonal anti-inflammatory drugs and other continuous use. Intravenous corticoid is the treatment of choice, particularly pulse therapy with methylprednisolone. Currently, the second medication indicated by the literature is cyclosporin, particularly in patients not responsive to corticosteroids [11].
Elevated levels of ferritin are important not only to differentiate patients with systemic JIA from other JIA, but also to identify those who may develop more severe disease and to monitor the possible development of MAS. Studies shown heterogeneity in patients with systemic JIA, and that group with highly ferritin being likely to complicate MAS [2].
After the correct diagnosis and appropriate treatment, the patient have been monitoring quarterly, showing not syndrome recurrence and not development of macrophagic activation syndrome.
Conclusion
Because it is a rare rheumatologic disease with a variable clinical condition, SJIA is diagnosis of exclusion, requiring careful and detailed investigation. In the presented case, the diagnosis was established after 18 days of the beginning of the clinical manifestations. The treatment was made based on non-steroidal anti-inflammatory and, later, steroidal anti-inflammatory and immunosuppressive drugs. Patient monitoring should be done continuously, as there is no consensus regarding the duration of treatment and the possibility of complications, such as macrophagic activation syndrome, which, if not adequately addressed and treated, may results in acute liver failure, disseminated intravascular coagulation, coma, and multiple organ failure.
References
- Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J (2005) Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med 201: 1479-1486. [Crossref]
- Guimarães JAC (2011) Pathogenesis of systemic juvenile idiopathic arthritis: role of innate immunity [master’s thesis]. Porto (PT): Porto University.
- Yokota S, Miyamae T, Imagawa T, Iwata N, Katakura S, et al. (2005) Therapeutic efficacy of humanized recombinant anti–interleukin-6 receptor antibody in children with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum 52: 818-825. [Crossref]
- Ferreira CS, Ribeiro EM, França BAA, Pignatti VTP, Freire M, et al. (2007) Síndrome de Evans na evolução da doença de Still. Rev Pediatr Moderna 43: 327-328.
- Carvalho RL, Tavares CM, Kuschnir MC, Aquino JHW (2007) Doença de Still na adolescência. Adolesc Saúde 4:53-55.
- Rosmaninho I, Guerra P, Guerra A, Brito I (2003) Artrite idiopática juvenil – forma sistêmica. Um caso particular. Acta Pediatr Port 34: 125-127.
- Ramos VCS, Ronchezel MV, Okuda EM, Sacchetti SB (2006) Caracterização epidemiológica, clínica e laboratorial de 100 crianças com artrite reumatóide juvenil. Rev Paul de Pediatr 24: 335-342.
- Bywaters EG (1971) Still's disease in the adult. Ann Rheum Dis 30: 121-133. [Crossref]
- Hadchouel M, Prieur AM, Griscelli C (1985) Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr 106: 561-566. [Crossref]
- Sen V, Ece A, Uluca U, Günes A, Yel S, et al. (2015) Evaluation of children with juvenile idiopathic arthritis in southeastern Turkey: a single center experience. Hippokratia 19: 63-68. [Crossref]
- Pasqualotto FS, Ramos MD, Moussalem GF, Fortunato M, Assis SB, et al. (2009) Ativação macrofágica (SAM) em um paciente de 14 anos. Rev Biodiv 8:78-79.