Abstract
The novel coronavirus SARS-CoV-2, responsible for COVID-19 pandemic, has triggered an unparalleled worldwide health crisis. The virus encoded main protease, Mpro is a promising drug target, and discovering its inhibitors is necessary to curtail such emerging viral infections. PDB has several co-crystal structures of Mpro with inhibitors. The inhibitors are broadly classified as covalent and non-covalent, where the latter class of inhibitors are safe and less reactive. We aimed to find non-covalent inhibitors of Mpro utilizing a library of natural product compounds by virtual screening and molecular docking re- scoring using two different algorithms. The top scored five compounds have better binding affinities against Mpro in nM to sub-nM ranges, than existing non-covalent inhibitors. They also meet the expected Lipinski and Veber drug-likeness standards and have acceptable ADME characteristics. Our analysis show that these compounds exhibit high binding affinity due to multiple hydrogen bonded interactions with the peptide backbone, oxyanion hole and catalytic dyad at the Mpro active site. Our study shows that the identified compounds, many of which are present in rhizomes, can inhibit Mpro and can be exploited to develop new non-covalent inhibitors with fewer adverse effects for effective treatment of SARS-CoV-2 infections.
Keywords
SARS-CoV-2 main protease, diaryl heptanoid, rhizome, binding affinity, non-covalent inhibitor, molecular docking, drug discover
Abbreviations
3CLpro: 3-chymotrypsin-like protease; Nsp: Non-structural protein; ORF: Open Reading Frame; PDB: Protein Data Bank; LE: Ligand Efficiency; NPC: Natural Product Compound
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for the Coronavirus disease 2019 (COVID-19) pandemic [1]. SARS-CoV-2 is highly contagious and causes severe respiratory illness and is responsible for more than 6.8 million deaths worldwide [2]. SARS-CoV- 2 are enveloped, positive-single stranded RNA viruses with a nucleocapsid, and the genomic size of 30 kb [3]. Upon entry into the host’s cells by binding of the SARS-CoV-2 spike or S protein (S1) to the ACE2 receptors which are abundantly present on alveolar epithelial cells [4]. The viral attachment process is followed by priming the spike protein S2 subunit by the host transmembrane serine protease 2 (TMPRSS2) that facilitates cell entry [4]. Replication of the viral RNA is initiated with the synthesis of polyprotein pp1a/pp1ab chain using its virally encoded polymerase RdRp and transcription occurs through the Replication-Transcription Complex (RTC) [5]. In the viral genome, six ORFs are present and a programmed ribosomal -1 frame shift between ORF1a and ORF1b, guides the production of two fusion proteins, pp1a and pp1ab [6]. Other ORFs encode structural proteins, including spike, membrane, envelope, and nucleocapsid proteins and accessory protein chains [7]. Both pp1a and pp1ab polypeptides are processed at 11 sites by virally encoded chymotrypsin-like protease (3CLpro) or main protease (Mpro) [8]. Currently, a variety of therapeutic options are available to the manage COVID-19 that include antiviral drugs targeting Mpro (paxlovid) [9] and RNA-dependent RNA polymerase (remdesivir and molnupiravir)
[10], anti-SARS-CoV-2 monoclonal antibodies (bamlanivimab/etesevimab, casirivimab/imdevimab, bebtelovimab, sotrovimab), anti-inflammatory drugs (dexamethasone) and immunomodulators (kineret, baricitinib and tocilizumab) [11].
SARS-CoV-2 encodes few enzymes, amongst them Nsp5 also called main protease (Mpro), is a 33.8 kDa cysteine protease and function as a dimer [12]. Mpro plays crucial role by cleaving the viral polyproteins, pp1a and pp1ab into functional proteins by cleaving at 11 unique sites with sequences LQ↓(S/A/G), generating the 12 functional proteins (NSPs 5-16) necessary for virus replication [13]. Thus functional importance of Mpro in the viral life cycle combined with the absence of close homologues in humans, identify Mpro as an attractive target for the design of antiviral drugs. As a result, the identification and development of potent covalent and non-covalent Mpro inhibitors is urgently required to combat SARS- CoV-2 infections and many antiviral drugs are being developed against Mpro [14].
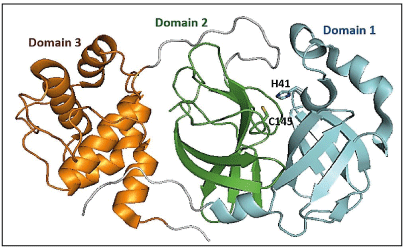
Crystal structures of SARS-CoV-2 Mpro show that the two protomers (306 residues) are non-covalently associated and form a functional homodimer. Each chain of protomer comprises of three domains (domains I, II, and III) (Figure 1a). Domains I (residues 8–101) and II (residues 102–184) are made up of six antiparallel β-barrels [12]. An antiparallel cluster of five α-helices forms domain III (residues 201– 303), which is connected to domain II via a long loop (residues 185–200). The active site cavity where substrate peptide binds, lies in the cleft between domains I and II, which has a Cys-His catalytic dyad involved in the proteolysis of substrates (Figure 1b) [15]. The substrate binding cavity contains a series of pocket (S4, S3, S2, S1, S1’, S2’, S3’, S4’), each one of them accommodates a single amino acid residue (P4, P3, P2, P1(Gln), P1’, P2’, P3’, P1’) of the substrate from N-terminus to C-terminus [16]. The substrate cavity is complete only after dimerization where the N-terminus finger residues 1-7 of other protomer interacts. Mpro inhibitors are broadly classified as covalent and non-covalent, where the non-covalent inhibitors are safe to use because of the presence of reactive groups present in covalent inhibitors. There are several co-crystal structures and reports on reversible and irreversible covalent inhibitors targeting the reactive cysteine, Cys145, and non-covalent inhibitors of Mpro [17-19]. Recently FDA approved a reversible inhibitor, nirmatrelvir against Mpro [20].
There is an urgent need to identify new Mpro inhibitors and there are no FDA approved non-covalent inhibitors against Mpro. Several non-covalent inhibitors targeting Mpro have been reported [21]. Natural products have emerged as a promising source of new drugs and most of the FDA approved drugs are based NPCs [10,22]. The rhizomes have traditionally been used for their medicinal properties and are an abundant source of bioactive compounds, including polyphenols, flavonoids, terpenoids, and alkaloids, that needs to be fully explored [23-26]. In our study, we explored the potential of Natural Product Compounds (NPCs) some of which are present in rhizomes, Alpinia officinarum, Curcuma longa and Zingiber officinale as a source of bioactive compounds. Many NPCs are known to be effective against viral infections [27-29]. Molecular docking has been an efficient tool for novel drug discovery by targeting proteins causing diseases. We used structure based virtual screening by molecular docking to identify NPCs that have the potential to inhibit Mpro. Autodock vina docking employs Broyden-Fletcher- Goldfarb-Shanno algorithm which improves the average accuracy of the binding mode predictions as compared to Autodock 4 [30,31]. FlexX employs an incremental construction algorithm which attempts to reconstruct the bound ligand by first placing a rigid anchor in the binding site and later using algorithm to add fragments and complete the ligand structure [32]. We aimed to identify non-covalent inhibitors of Mpro from a library of NPCs, some of them are present in rhizomes, by virtual screening using molecular docking and re-scoring using two different algorithms. An in-house library of compounds was prepared targeting the Mpro active site. We present the results of our virtual screening studies using Autodock vina and top scored compounds were identified after re-scoring using Hyde FlexX [33]. The top scored docked compounds belonging to diaryl heptanoids, show highest binding affinity to Mpro due to multiple hydrogen bonds near the catalytic residues. Five top scored compounds have excellent binding affinity, and are compliant with the predicted Lipinski [34] and Veber [35] rules for drug-likeness and good ADMET properties [36]. The results of our study demonstrates that the identified compounds can inhibit Mpro, and these can be used for further optimizations, in to develop new non-covalent inhibitors for effective treatment of SARS-CoV-2 infections.
Materials and Methods
Ligand and Protein Preparation and Molecular Docking
A library of more than hundred NPCs, some of which are found in rhizomes, Alpinia officinarum, Curcuma longa and Zingiber officinale (ginger), was prepared to identify molecules that could potentially bind to and inhibit Mpro. The 3D structures compounds with hydrogen atoms were generated using ACD Chemsketch 2021.2.1 [37] and energy minimized atomic coordinates were saved. The file format conversions to 3D sdf or PDBQT were done by Openbabel [38]. Crystal structure of Mpro (PDB ID: 6Y2F) was downloaded from Protein Data Bank (PDB) [39]. The protein PDBQT was prepared using Graphical User Interface program Autodock tools (ADT) and was assigned polar hydrogens, united atom Kollman charges, solvation parameters and fragmental volumes to the protein [31]. A three-dimensional grid, encompassing the receptor substrate binding pocket, was defined with center at (X, Y, Z) (10.2Å, 1.6Å, 21.8Å) and box dimensions (X, Y, Z) (30Å, 30Å, 30Å). The prepared molecules were docked against Mpro using Autodock vina with an exhaustiveness of 10 employing iterative local search global optimizer [30]. AutoDock vina generated ten different conformations for each ligand. All the compounds were docked using the same docking parameters to generate log and output files containing binding energies of compound with Mpro. Generated output poses are viewed using AutoDock tools, Pymol [40] and Biovia [41]. The pose with lowest energy of binding was extracted and aligned with receptor structure for further analysis. The ligands having less than -6.5 kcal/mol binding affinities were re-ranked using FlexX [32]. The same PDB file was used during docking in FlexX, and the crystallographic ligand was defined as the docking site. The docking pocket of the enzyme was defined around the bound reference ligand having a radius of 9Å. The maximum number of poses was set to 200 and top 10 poses were extracted and saved. Hyde calculations were done on each output pose [33]. The docked poses were analyzed using in-built visual interfaces, FlexX, Pymol and Biovia for further analysis and making figures. The top five docked compounds were analyzed for predicting their drug-likeness and ADME (absorption, distribution, metabolism and excretion) properties using Swiss ADME [36].
Results
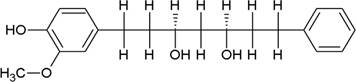
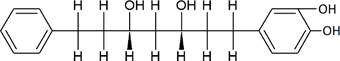
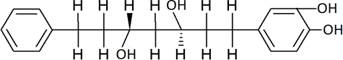
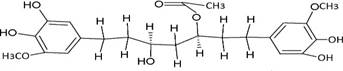
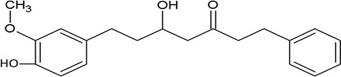
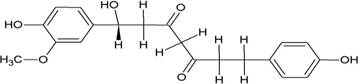
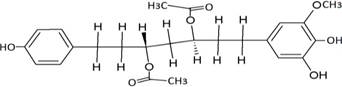
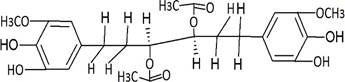
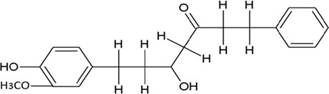
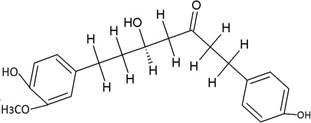
The structures of the ten best ligands, having highest Gibbs free energy of binding (kJ/mol) using Hyde, are listed in Table 1. Our docking results show that the diaryl heptanoid class of compounds exhibit best binding affinities against Mpro. The size of these compounds fit in the Mpro binding cleft. The Hyde docking energy was calculated by addition of van der Waals force, bond energy, dissolve energy, electrostatic energy and internal energy [33]. The poses of each ligand were analyzed in the binding site for hydrogen bonded and van der Waals interactions and visualized by PyMOL v2.0 viewer [40]. These compounds were selected having free energy of binding less than -30 kJ/mol and Ligand Efficiency (LE) less than 0.21. Lower free energy of binding denotes lower binding affinity or stronger protein-ligand binding. The higher LE values denotes each ligand atom has favourable interactions with protein. Adg5 binds to Mpro exhibiting highest Hyde binding affinity of −45.7 kJ/mol and LE of 0.39 followed closely by adg4 and adg3 shown in Table 1. Adg31 has additional hydroxyl and methoxyl groups and has similar affinity but lower LE value 0.28 than the top three compounds. The four compounds have 1-10 nM binding affinities. Adg53 and adg46 have lower than -40 kJ/mol binding affinities within 100nM range and higher that 0.30 LE values. Adg28 and other compounds have lower LE values and affinities lying between -40 kJ/mol to -36 kJ/mol (100 nM-1 µM range).
Interactions in the Main Protease active site
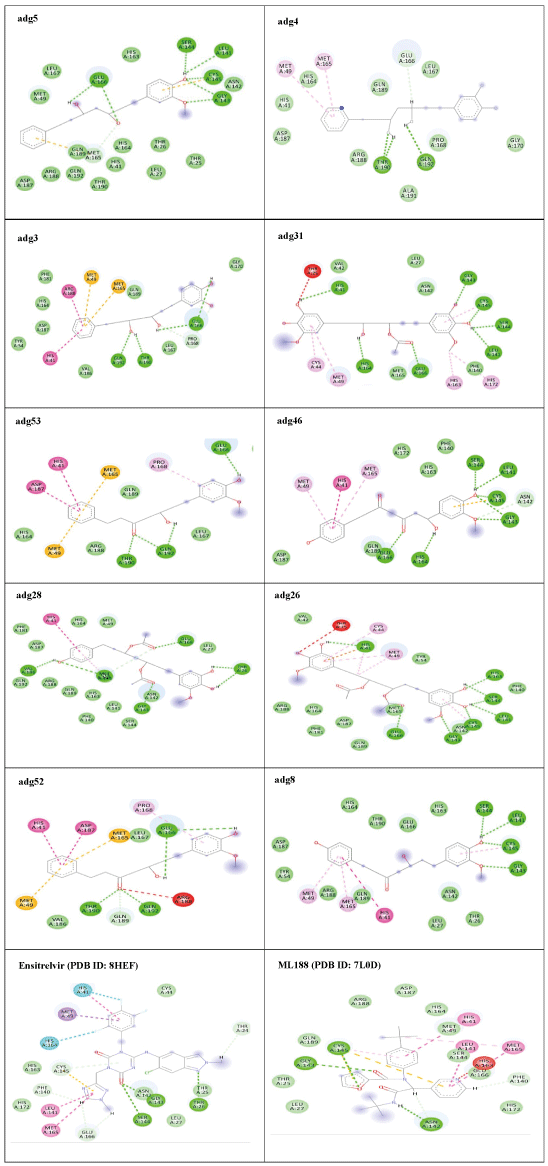
The ligand binding cleft of Mpro is long and lies on the surface having volume of 720 Ǻ3 [42]. Figure 2 panel shows best docked poses of our top ten compounds in active site cavity on Mpro surface. The docked compounds lie within this volume (Figure 2).
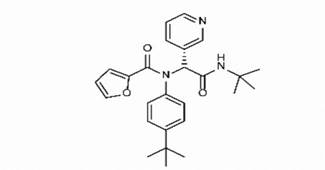
Adg5 makes seven hydrogen bonds with amino acid residues Ser144, Leu141, Glu166, Gly143 and Cys145 shown in Figure 3 and six of them are with peptide backbone. The compound adg5 has highest affinity due to hydrogen bonds with one bifurcated hydrogen bond. It interacts with catalytic Cys145 and Met145 through pi-sulphur interactions. The amino acid residues Met49, Leu167, His143, Phe140, Phe181, Asp187, Arg188, Gln192, Gln189, Val186, Thr190, His41, His164, Thr26, Leu27 and Thr25 interact with adg5 through van der Waals interactions. Adg4 and adg3 makes four hydrogen bonds with bifurcations with amino acid residues Gln192, Thr190 and Glu166 shown in Figure 3. The amino acid residues His164, Gln189, Gly170, Phe181, Arg188, Val186 and Leu167 interact with adg4 via van der Waals interactions. Adg31 forms many bifurcated hydrogen bonds with Cys44, Thr25, His41, His164, Gly143, Leu141, Ser144 Cys145 and Glu166 amino acid residues and most of them are with peptide backbone. It has van der Waals interactions with His164, Gln189, Gly170, Phe181, Arg188, Val186 and Leu167. Adg53 makes three hydrogen bonds with Thr190, Gln192 and Glu166 and interacts with catalytic residue His41 and Asp187 through pi interactions. It also forms pi-sulphur interactions with Met165 and Met49. The hydrogen bonds and intermolecular interactions between the rest of the compounds with Mpro are shown in Figure 3. Many Mpro residues interacting with the inhibitors are conserved residues and have crucial role in catalysis. Two noncovalent ligands, ensitrelvir and ML188 complexed with Mpro were docked in the active site of Mpro, and results are put in last two rows in tables. Mpro makes few hydrogen bonds with ensitrelvir through Ser144, Gly 143 and Thr26. ML188 makes hydrogen bonds with Gly143 and Asn142 residues of Mpro.
Discussions
SARS-CoV-2 has many druggable targets, amongst which Mpro is a promising candidate. Mpro has less off-target effects because of its glutamine (Gln) cleavage recognition site at P1 substrate site is unique, and has not been not observed in any human protease. Mpro has well defined active site with few conserved residues. The substrate and ligand binding pocket of Mpro lie on the surface having volume of 720 Ǻ3 [43]. The active site of Mpro is highly conserved among all SARS-CoV variants and is composed of four subsite pockets (S1’, S1, S2, S2’, S3 and S3’), which can easily accommodate a wide selection of inhibitors and fragments. The cleavage site of substrate, S1-S1’ flanks the catalytic dyad Cys145-His41 residues. Thus, the inhibitors should be large enough to occupy the pockets and properly functionalized to improve their interactions with Mpro side chain and peptide backbone residues lying in these pockets. Plant-based potential bioactive compounds, having antiviral properties have been proved superior, and could be combined with the preexisting therapies, along with the different delivery methods to enhance the effectiveness of antiviral along with good bioavailability [24,43]. Many natural products have been in clinical trials to treat HIV and HCV [44-46]. Rhizomes are underground stems that are rich in a variety of compounds which have been shown to have antiviral activity in vitro [28-29]. The rhizomes, Alpinia officinarum, Curcuma longa and Zingiber officinale (ginger) are of particular interest because they contain a variety of biologically active compounds that have been shown to have antiviral activity [27,29]. We found the compounds belonging to diaryl heptanoids class bind within the pocket. The top five compounds having binding energies in the nM range were analysed for druggability and ADMET properties which are shown in Table 2.
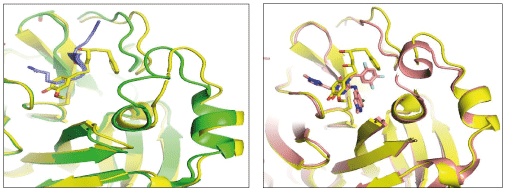
The hydrogen bonds between hydrophilic groups of the compounds, and side and main chains atoms of Mpro are shown in Figure 3 panel. The hydrogen bond lengths lie between 2.7 Å to 3.2 Å, and they contribute significantly to the free energies of binding with Mpro. The longer distance between the catalytic dyad residues, as compared to serine proteases, allows for closer contact of these non-peptide inhibitors with the catalytic residues. These compounds interact directly with the catalytic dyad, the oxyanion hole and peptide backbone in catalytic site through hydrogen bonds. Several Mpro drug-resistant mutants against recent FDA approved drug, nirmaltevir have been predicted [47]. Many circulating natural Mpro variants are also emerging which is evident from more than 15 million SARS-CoV-2 genome sequences deposited [https://gisaid.org/]. The hydrogen bonding of the compounds with peptide backbone and conserved residues of Mpro has important implications because these compounds will be least affected by emerging drug-resistant Mpro mutants and circulating natural Mpro variants. In Figure 4, we show one of the docked compound, adg5 is compared with co-crystallized inhibitor, Ensitrelvir and substrate peptide, where the crystal structures of Mpro complexes are superposed with each other. The hydroxyl or methoxy containing ring of the docked compounds superpose well with the central ring of the inhibitor near catalytic Cys145, and the docked compound backbone superpose close to the crystallized C-terminal substrate peptide backbone spanning P1’-P3’residues. Thus our docked compounds bind to Mpro in similar orientation to the potent inhibitors in the active site of Mpro.
The druggability and ADME predictions are shown in Table 2. The spider plots of the compounds shown in last row of Table 2, indicate that all five compounds have favourable predicted values of the parameters. Adg31 shows slight deviations on flexibility and polar parameters. These compounds have acceptable Lipinksy, Ghose and Veber parameters. Adg5 and adg53 have no PAINS alert whereas adg4, adg3 and adg31 has only one PAINS alert. From the analysis of structures of crystallized inhibitors with Mpro deposited in PDB, non-covalent inhibitors are generally smaller in size (<MW> ~ 348 ± 35% g/mol). The identified compounds have similar molecular weights. The predictions also indicate that all five have good synthetic accessibility values below 4 and are inhibitors of human P450 cytochromes. Hence these compounds can be potential drug candidates.
Our molecular docking results indicate the diarylheptanoids class of compounds, some of which are present in rhizomes of Alpinia officinarum and Zingiber officinale (ginger), bind with high in-silico affinity (nM range) to SARS-CoV-2 Mpro. The identified five compounds bind to Mpro having multiple interactions, especially hydrogen bonds with protein backbone and interactions with highly conserved residues of Mpro, and hence are expected to be less susceptible to emerging drug-resistant and naturally circulating Mpro mutations. They also have favourable ADME properties and hence can be used to develop anti-SARS-CoV-2 inhibitors and formulations to control the viral infections since the rhizomes are already being consumed. The identified compounds will be a good start point to develop non-covalent inhibitors against Mpro with less side effects as compared to covalent inhibitors. Also they provide a skeletal know how to the synthetic chemists to further develop them as more potent anti-SARS-CoV-2 drugs. Few of these compounds are being synthesized in our laboratory, and further in vitro studies are planned. The inhibitors can be lead novel non-covalent Mpro inhibitors, which can be used alone or in combination with FDA approved MPro inhibitor, nirmaltevir and RdRp inhibitors.
Conclusions
The SARS-CoV-2 Mpro is an important drug target due to its essential role in viral replication. Our virtual screening studies identified few diaryl heptanoid compounds from Alpinia officinarum and Zingiber officinale (ginger). They bind to Mpro with nM affinities having hydrogen bonds with protein backbone, and interact with Mpro’s highly conserved residues. The best five docked molecules bind to Mpro with better affinities than the two non-covalent co-crystallized inhibitors. The docked compounds bind in similar orientation to ensitrelvir and the substrate co-crystallized with Mpro. These compounds are predicted to have good ADME properties and are less susceptible to emerging drug-resistant and circulating Mpro mutations. Our structure based molecular docking analysis demonstrates that these inhibitors and related compounds are good non-covalent inhibitors candidates, which will lead to the development of new non-covalent Mpro inhibitors with fewer side effects.
Acknowledgements
Authors are grateful to Bhabha Atomic Research Centre, Department of Atomic Energy, India for support.
Scoring Function. J Comput Aided Mol Des. 27 (1): 15–29. https://doi.org/10.1007/s10822-012- 9626-2.