Abstract
ALK-positive Non-Small-Cell Lung Cancer (NSCLC) is a defined subgroup of lung cancer. Crizotinib was the first ALK-inhibitor introduced in clinical practice after two phase III trials demonstrated its superiority over chemotherapy both in second and in first line of treatment. Approximately within ten months patients developed acquired resistance to crizotinib and relapse. Second and third generation ALK-inhibitors are more potent molecules designed to overcome crizotinib resistance. Ceritinib, alectinib and brigatinib are approved by FDA as subsequent therapy in patients who have progressed after crizotinib. Lorlatinib and entrectinib are in different phases of clinical development. Moreover some of these agents are compared to crizotinib in first line setting to evaluate if an upfront more potent inhibitor could control disease longer than a sequential strategy. Despite the efficacy of second-generation ALK inhibitors, patients relapse. Each ALK-inhibitor is characterized by a distinct resistence profile with important clinical consequences in the choice of subsequent therapy.
Key words
non-small-cell lung cancer (NSCLC), EML4-ALK rearrangement, crizotinib, anaplastic lymphoma kinase inhibitors (ALK inhibitors)
Introduction
Unselected patients with metastatic Non-Small-Cell Lung Cancer (NSCLC) treated with conventional chemotherapy present a median survival of 10-12 months. According to the oncogene-addiction paradigm, the inhibition of molecular drivers by target agents could reduce tumor burden and improve patient survival [1,2]. Oncogenic ALK gene rearrangements are one of the several molecular alterations described in NSCLC especially in adenocarcinoma. Other molecular drivers are sensitizing EGFR gene mutations, ROS1 gene rearrangements, BRAF V600E target mutations. Emerging biomarkers, for which targeted agents are under investgation, include HER-2 mutations, RET gene rearrangements, high-level MET amplification or MET exon 14 skipping mutations (METex14) [3]. ALK is an insulin receptor tyrosine kinase with unclear physiologic functions. In humans, ALK expression is limited to the adult brain, no expression has been evidenced in normal lung tissue [4]. ALK gene rearrangements, occurring approximately in 4% of lung adenocarcinoma, define a distinct molecular subtype of NSCLC. The diagnosis of this gene alteration, although rare, offers patients the opportunity to receive highly effective target therapy [5]. Chromosomal rearrangements involving ALK gene were described in Non-small cell lung cancer (NSCLC), anaplastic large cell lymphoma (ALCL), and inflammatory myofibroblastic tumor (IMT). These rearrangements lead to the expression of ALK fusion genes in which the fusion partner mediates ligand-dependent oligomerization of ALK, resulting in constitutive ALK kinase activation [6-18]. In NSCLC the major fusion partner is echinoderm-microtubule-associated protein-like 4 (EML4), with the formation of the EML-ALK fusion protein. More than other 20 ALK fusion partners have been identified in lung cancer but the clinical significance of these fusion protein requires further investigations [7]. ALK rearrangements are mostly found in non-squamous lung histology, never- or light smokers and in younger patients. These clinical-pathologic features should not be utilized in selecting patients for testing [3]. Therefore ALK testing is recommended for patients with adenocarcinoma, for lung cancer of mix histology with an adenocarcinoma component, for limited specimens such as biopsy and cytology specimens where adenocarcinoma component cannot be completely excluded and for never-smokers who are younger than 50 years and have tumor of squamous histology [3,5-8]. ALK IHC assays are validated, standardized and cost-effective screening method to detect ALK rearrangement to select ALK-positive NSCLC . FISH can be use to confirm ALK positivity detected by an IHC assay. A practical cutoff value of 15% has been established to discriminate ALK-rearranged and ALK wild type NSCLCs [3,9-11].
Crizotinib
Crizotinib, a selected, first generation tyrosine kinase inhibitor (TKI) of ALK, ROS1 and MET, was the first ALK inhibitor introduced into clinical practice. The first phase I clinical trial of crizotinib (PROFILE 1001) was initially designed to test the activity of crizotinib in patients with MET deregulation (MET amplification or MET mutation). A significant tumor shrinkage was observed in patients with MET-amplification and in patients with NSCLC harboring ALK rearrangement. So the protocol was amended to screen simultaneously patients for both ALK translocation and MET amplification. In ALK-positive NSCLC updated results showed a response rate (RR) of more than 60%, a median progression free survival (PFS) of 9 months [9,12]. Similar findings have been observed in the phase II study PROFILE 1005 [13]. Two phase III studies highlighted the advantage of crizotinib over standard chemotherapy [14,15]. In the PROFILE 1007 study, second-line crizotinib was compared to standard chemotherapy (pemetrexed or docetaxel) in patients with advanced ALK-positive NSCLC progressing after one prior platinum-based chemotherapy. Crizotinib was associated with a RR of 65% and a median PFS of 7.7 months while chemotherapy showed a RR of 20% and a median PFS of 3.0 months [14]. The PROFILE 1014 trial demonstrated improvement in PFS of crizotinib over standard platinum-based chemotherapy in first-line advanced non-squamous ALK-positive NSCLC. In the crizotinib group median PFS was 10.9 months, RR was 74%, in chemotherapy arm PFS was 7.0 and RR was 45% [15]. In both studies the improvement in PFS did not translated into an advantage in overall survival (OS) because of confounding effect of cross-over. Based on these data crizotinib was approved both as first-line and as subsequent therapy in patients with ALK-positive NSCLC [3].
Resistance to crizotinib
After a median time of 10 months, patients become refractory to crizotinib and relapse on therapy [2,3,5-17]. A deeper understanding of molecular processes of acquired crizotinib resistance results from analysis of post-progression biopsy specimens. Central nervous system (CNS) is the first site of progression in approximately 50% of patients, suggesting inadequate penetration into the CNS by crizotinib [18-20]. Mechanisms responsible of acquired resistance are target-dependent (50%), non target-dependent (30%) and unknown (20%) (Figure 1). The main mechanisms of acquired resistance are: genetic alteration of the drug-target (point mutation and/or gene amplification) and activation of bypass signaling with the activation of a parallel pathway obviating the need for the drug [17]. Target-dependent mechanisms preserve the domain of ALK signaling and can occur through mutations in the kinase domain. The most common ALK resistance mutations were the gatekeeper L1196M substitution (which is analogous to T790M in epidermal growth factor receptor) [17] and G1269A. These alterations were present in only 7% and 4% of all of the crizotinib-resistant specimens, respectively. The remaining ALK resistance mutations included: C1156Y (2%), G1202R (2%), I1171T (2%), S1206Y (2%), and E1210K (2%) [2,16] (Figure 2). Another mechanism of acquired resistance to crizotinib is an increase or amplification of the number of rearranged echinoderm microtubule-associated protein-like 4 (EML)-ALK genes. Therefore not all EML-ALK fusion proteins are inhibited by clinically achievable doses of crizotinib [2,17]. Non target dependent acquired resistance involved the activation of a parallel or bypass signaling pathways obviating the need for the original drug. Epidermal growth factor receptor (EGFR) activation, KIT amplification, KRAS mutations may mediate acquired crizotinib resistance [17] although their role is unclear [17]. ALK-rearranged-NSCLC treated with crizotinib might develop KRAS and EGFR mutated ALK-negative tumors [21]. Heat shock protein 90 (Hsp90) is a chaperone protein assisting other proteins in proper folding, stability and function of several molecules some of which involved in tumor growth. ALK fusion proteins are Hsp 90 clients and Hsp90 inhibitors have shown activity against EML4-ALK in clinical and preclinical studies suggesting their use in crizotinib-resistance ALK-positive NSCLC [17,22-24]. Preclinical studies highlight that Epithelial-Mesenchymal Transition (EMT), which enhances cell motility and invasiveness [25], is associated with crizotinib resistance [26-28]. Transformation to Small-Cell Lung Cancer (SCLC) is a mechanism of target-independent acquired resistance in EGFR-mutation-positive NSCLC which is also rarely described in ALK-rearranged lung cancer [29-31]. Presumably in patients experiencing slow progression, in limited pre-existing sites, or in a single new site without worsening of clinical status, the main mechanism of resistance is ALK-dominant. In this condition the discontinuation of ALK inhibition is associated with the risk of disease flare [32,33]. Continuation of crizotinib beyond progression in association with local therapies (radiotherapy, local ablation or surgery against only the sites of disease progression) may extend disease control by more than 6 months [34]. In 120 patients enrolled in PROFILE 1001 and 1005 who continued crizotinib for >3 weeks post-RECIST progression, the median duration of crizotinib treatment beyond progression was 19.4 weeks and median OS from the time of first progression was significantly longer for patients continuing crizotinib compared with patients who stopped [35]. Patients selection is essential because subjects who benefited from continuing crizotinib have a good performance status, had achieved an objective response to crizotinib, and had a site of progressive disease that was manageable with local therapy [36]. Patients undergoing rapid radiological progression and impairing in clinical conditions have become completely refractory to crizotinib or addicted to another driver. Crizotinib should therefore be replaced by conventional chemotherapy or a next generation ALK inhibitor [35]. Primary resistance is the lack of response to ALK-inhibitors. Mechanisms underling primary resistance are not clearly defined [2].
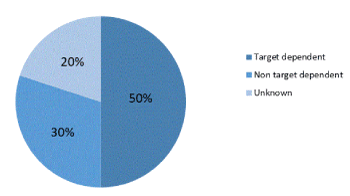
Figure 1. Mechanisms underlying acquired resistance to crizotinib [2]
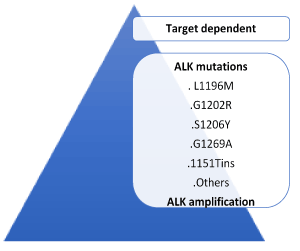
Figure 2. Target-dependent acquired resistance to crizotinib [2]
Second generation ALK inhibitors
Second- and third-generation ALK-inhibitors, developed to overcome acquired crizotinib resistance, have been investigated in clinical trials both in crizotinib-refractory and in crizotinib-naïve settings.
Ceritinib
Ceritinib (LDK378) is a second generation ALK and ROS1 inhibitor [37]. ASCEND-1 is a phase I study aimed to assess activity of ceritinib in both ALK inhibitor-pretreated and ALK inhibitor-naive patients with ALK-rearranged NSCLC. An overall response was reported in 72% of 83 ALK inhibitor-naive patients and in 56% of 163 ALK inhibitor-pretreated patients. Median duration of response was 17.0 months in ALK inhibitor-naive patients and 8.3 months in ALK inhibitor-pretreated patients. Median progression-free survival was 18.4 months in ALK inhibitor-naive patients and 6.9 months in ALK inhibitor-pretreated patients [38]. Based on this study, ceritinib was approved by FDA in ALK-positive NSCLC who have progressed on crizotinib [3,38]. ASCEND-2 is a phase II study evaluated efficacy and safety of ceritinib in ALK-positive NSCLC previously treated with chemotherapy and crizotinib. Patients should have received cytotoxic therapy (1–3 lines, including 1 platinum doublet) and progressed on crizotinib as the last treatment prior to study entry. The overall response rate (ORR) by investigators was 38.6% (35.7% by blinded independent review committee), with a PFS of 5.7 months [39]. ASCEND-3 is a single arm phase II study of ceritinib in ALK-inhibitor naive patients in ALK-positive NSCLC. 98.4% of patients had received at least 1 line of prior chemotherapy and 25% of patients had received ≥3 prior antineoplastic regimens. Whole-body ORR of 63.7% (by blinded independent review committee of 58.9%), median PFS was 11.1 months with a median follow-up of 8.3 months [40].ASCEND-4 is phase 3 study in untreated patients with stage IIIB/IV ALK-rearranged non-squamous NSCLC randomized to receive ceritinib or platinum-based chemotherapy. Median progression-free survival (assessed by blinded independent review committee) was 16.6 months in the ceritinib group and 8.1 months in the chemotherapy group [41]. Based on this phase III trial FDA approved ceritinib as first-line therapy for patient with ALK-positive metastatic NSCLC [3]. ASCEND-5 is a phase III trial aimed to assess efficacy and safety of ceritinib versus single-agent chemotherapy (pemetrexed or docetaxel) in patients with advanced ALK-rearranged NSCLC who had previously progressed following crizotinib and platinum-based doublet chemotherapy. Patients treated with ceritinib showed a significant improvement in median progression-free survival compared with chemotherapy (5.4 months for ceritinib vs 1.6 months for chemotherapy [42].
Alectinib
Alectinib (CH5424802/RO5424802) is an oral TKI inhibitor of ALK and RET rearrangements which is now recommended as the preferred first-line therapy in patients with previously untreated advanced ALK-positive NSCLC based on results of ALEX and J-ALEX trial [3]. ALEX trial is a randomized, open-label, phase 3 trial comparing alectinib with crizotinib in patients with previously untreated, advanced ALK-positive NSCLC, including those with asymptomatic brain or leptomeningeal metastases. Investigator assessed progression-free survival was 68.4% with alectinib and 48.7% with crizotinib. The median progression-free survival with alectinib was not reached as compared with 11.1 months with crizotinib. Independent review committee–assessed progression-free survival was also significantly longer with alectinib than with crizotinib (median progression free survival was 25.7 months and 10.4 months respectively). The time to CNS progression was significantly longer with alectinib than with crizotinib in the intention-to-treat population: 12% in the alectinib group had an event of CNS progression, as compared with 45% in the crizotinib group. Median duration of response was not estimable with alectinib and 11.1 months with crizotinb. Among patients with measurable CNS lesions CNS response rate was 81% in alectinib group and 50% in crizotinib group. The median duration of intracranial response was 17.3 months for alectinib and 5.5 months for crizotinib. The 12-month survival rate was 84.3% with alectinib and 82.5% with crizotinib. Overall survival data are currently immature. Alectinib demonstrated an intersting safety profile: despite the longer duration of treatment with alectinib (median 17.9 months vs 10.7 months with crizotinib), grade 3 to 5 adverse events occurred in 41% of the patients treated with alectinib and 50% of the patients treated with crizotinib [43]. The results of this trial are supported by those of the J-ALEX trial involving Japanese patients with ALK-positive previously untreated advanced NSCLC. Median PFS had not yet been reached with alectinib versus 10.2 months with crizotinib. Grade 3 or 4 adverse events were less frequent with alectinib when compared with crizotinib [44]. Alectinib is also approved by FDA for ALK-positive NSCLC who have progressed on are intolerant to crizotinib [45]. Shaw et al. evaluated alectinib in 87 patients with ALK-positive crizotinib-resistant NSCLC. ORR was 48%, median Duration Of Response (DOR) was 13.5 months [46]. In 138 patients who have progressed on crizotinib Ou et al. demonstrated a response rate of 50% and a median duration of response of 11.2 months [47]. In both trials alectinib showed an impressive activity in control of central nervous system (CNS) disease and a good safety profile with most adverse events of grade 1 or 2 [46,47].
Brigatinib
Brigatinib (AP26113) is a potent and selective second generation ALK and ROS1 inhibitor designed to overcome first generation crizotinib resistance. In preclinical models it showed activity against mutant form of EGFR [35]. Brigatinib displays superior in vitro and in vivo potency in NSCLC models compared with crizotinib: in ALK-positive cell lines and in xenograft mouse models brigatinib inhibited native ALK with 12-fold greater potency than crizotinib. Several secondary mutations at 11 different amino acid residues in ALK-sequence (G1123, T1151, L1152, C1156, I1171, F1174, L1196, G1202, D1203, S1206, and G1269), have been associated with clinical resistance to crizotinib and/or the second-generation ALK inhibitors ceritinib and alectinib. Brigatinib is a more potent inhibitor of native EML4-ALK than crizotinib, ceritinib, and alectinib and exhibits substantial activity against all secondary ALK mutations at clinically achievable concentrations. In vivo Brigatinib demonstrates antitumor activity against L1196M, the most common mutation associated with resistance to crizotinib, and G1202R which is associated with clinical resistance to crizotinib, ceritinib, and alectinib [48]. In April 2017, based on the results from ALTA trial, brigatinib received accelerated approval for the treatment of patients with ALK-positive metastatic NSCLC who have progressed on or are intolerant to crizotinib [3]. ALTA is a prospective, open-label, randomized, phase II trial assessing brigatinib efficacy and safety at 90 mg once daily (arm A) and 180 mg once daily with a 7-day lead-in at 90 mg (arm B) in 222 patients with crizotinib-refractory advanced ALK-positive NSCLC. Objective response rate (ORR) was 45% in arm A and 54% in arm B. In patients with measurable brain metastases the intracranical overall response rate was 42% and 67% respectively. Median progression-free survival was 9.2 months and 12.9 months in arm A and B respectively [49]. ALTA-1L study (ALK in lung cancer trial of brigatinib in first-line), is an ongoing phase III, randomized, open-label trial evaluating efficacy and safety of brigatinib versus crizotinib in ALK-positive locally advanced or metastatic NSCLC who have not previously been treated with an ALK inhibitor [50]. At first interim analysis progression-free survival was significantly longer in brigatinib than in crizotinib arm. At a median follow up of 11 months the median progression-free survival was not reached in brigatinib arm and 9.8 months in crizotinib arm. The one-year PFS was 67% in patients receiving brigatinib and 43% in patients receiving crizotinib. The objective response rate was 71% with brigatinib treatment compared to 60% with crizotinib. There was a clear benefit for brigatinib across all prespecified patient subgroups including patients with brain metastases. In patients with measurable brain metastases at baseline the confirmed ORR was 78% with brigatinib and 29% with crizotinib [74].
Lorlatinib
Lorlatinib (PF-06463922) is a third generation, reversible, ATP-competitive inhibitor of ALK and ROS1. In the ALK tyrosine kinase domain lorlatinib selectively inhibits leucine at position 1195 (L1195). Lorlatinib has shown superior potency compared with clinically available ALK inhibitors and it also addresses the two major mechanisms of clinical relapse: ALK resistance mutations and brain metastasis [51]. In biochemical studies lorlatinib has proven to be more potent than crizotinib, ceritinib and alectinib against wild type ALK. It is also the most potent inhibitor against all clinically relevant crizotinib-, certinib- and alectinib-resistant ALK mutations [51]. It showed activity against L1196M and G1269A, two of the most frequently detected crizotinib-resistant mutations observed in clinical practice [51-53]. Moreover PF-06463922 inhibits 1151Tins and G1202R ALK mutants that confer a high-level of resistance to all second generation ALK inhibitors [51,54,55]. Lorlatinib is the most potent and brain penetrable ALK-Inhibitor designed to efficiently penetrate the blood barrier brain. Crizotinib is active against brain metastases but CNS is a common site of relapse [18-20]. Probably resistance is related to the inability of the drug to achieve therapeutic concentration in CNS compartment because of its high efflux by P glycoprotein (PGP) [56]. Second generation ALK inhibitors have shown moderate CNS activity [57]. Ceritinb is a PGP substrate and has limited brain penetration [58], alectinib is not a PGP substrate [57]. Patients treated with either ceritinib and alectinb relapse with brain metastases. Therefore it was necessary to design a more potent inhibitor which is not a substrate of PGP, which efficently penetrates the blood brain barrier with an increased CNS availability [51]. Patient with metastatic ALK-rearranged lung cancer received multiple ALK inhibitors during treatment course, including first-, second-, and third-generation inhibitors. Shaw et al. described resensitization to crizotinib after acquired resistance to lorlatinib. A patient with ALK-rearranged lung cancer, developed acquired resistance to crizotinib because of the substitution of cysteine by tyrosine at amino acid residue 1156 (C1156Y) in the ALK kinase domain. The disease did not respond to second generation ALK-inhibitors, but it responds to lorlatinib. When the tumor relapsed, sequencing of the resistant tumor revealed an ALK L1198F mutation in addition to the C1156Y mutation. The L1198F substitution confers resistance to lorlatinib but enhances binding to crizotinib resensitizing resistant cancers to crizotinib, a less potent and less selective first-generation inhibitor [59]. In a first-in man, dose-escalation phase I study, lorlatinib showed both systemic and intracranial activity in patients with advanced ALK-positive or ROS1-positive NSCLC, most of whom had CNS metastases and had previously been treated with two or more TKIs [60]. Lorlatinib might be an effective therapeutic strategy for patients with ALK-positive NSCLC who have become resistant to currently available TKIs, including second-generation ALK TKIs. A phase 3 trial is ongoing to to demonstrate whether lorlatinib is superior to crizotinib in advanced, treatment-naive, ALK-positive NSCLC [61].
Entrectinib
Entrectinib (RXDX-101) is a small molecule which inhibits tropomyosin-related kinase (TRK) TRKA, TRKB, TRKC, ROS1 and ALK rearrangements. Clinical activity of entrectinib has been assessed in 4 clinical trials [62-64]. Combined results derive from two phase 1 basket trials (ALKA-372-001 and STARTRK-1) conducted in 119 patients with advanced solid tumor harbouring a recurrent gene fusion involving NTRK1/2/3, ROS1, or ALK. Entrectinib demonstrated interesting antitumor activity in TKI-naïve patients harboring gene rearrangements involving NTRK, ROS1, or ALK genes. Clinical benefits were observed abroad a range of solid tumors regardless of histology, particularly in patients with NTRK-rearranged tumors. No responses were observed in patients with recurrent gene rearrangements previously treated with ROS1 or ALK inhibitors. Further investigation will be required to determine the activity of this drug in TKI pre-treated patients considering that RXDX-101 has shown preclinical activity against potential resistance mutations such as the ALK L1196M mutation that can emerge after crizotinib therapy in ALK-rearranged lung cancers. Entrectinib proved interesting intracranial activity against both metastatic disease and primary brain tumors [62]. STARTRK-2 is an ongoing phase 2 basket trial of entrectinib for the treatment of patients with solid tumors that harbor an NTRK1/2/3, ROS1, or ALK gene fusion designed to confirm the results of STARTRK-1 and ALKA [62,63]. The STARTRK-NG trial is a Phase 1/1b study of entrectinib in pediatric patients with cancer, including primary brain tumors, neuroblastoma, and other non-neuroblastoma, extracranial solid tumors harboring NTRK, ROS1, or ALK gene fusions [64].
Resistance to second generation ALK inhibitors
Despite the efficacy of second-generation ALK inhibitors, patients invariably relapse. While only 20% of ALK-positive patients developed ALK resistance mutations on crizotinib, almost 56% of patients progressing on second-generation ALK inhibitor developed ALK resistance mutations (ceritinib 54%, alectinib 53%, and brigatinib 71%) reflecting the greater potency and selectivity of these agents compared with crizotinib (Table 1) [65]. ALK G1202R is the most common ALK resistance mutation after treatment with second-generation ALK inhibitor. The spectrum of other ALK resistance mutations differs across agents. Each ALK inhibitor is associated with a distinct range of ALK resistance mutations which provides differential sensitivities to second-generation ALK inhibitors with important clinical implications. Resistance profiles may evolve over time and in response to sequential ALK inhibitors, particular ALK resistance mutations inform the choice of subsequent ALK targeted therapies, especially after failure of two ALK inhibitors. Lorlatinib has shown to be active against all single ALK resistance mutations and was the only ALK inhibitor with significant activity against ALK G1202R. Compound resistance mutations are principally described in patients who had received multiple ALK inhibitors, suggesting that their development may be facilitate by sequential use of ALK inhibitors. Compound resistance mutations in ALK-positive NSCLC are analogous to drug resistant T790M/C797S described following sequential treatment with first- and third-generation EGFR inhibitors in EGFR-mutant NSCLC.
Table 1. Frequence of ALK-resistance mutations after first and second generation ALK-inhibitors [66]
ALK Resistance
Mutations |
Crizotinib
(N=55) |
Ceritinib
(N=24) |
Alectinib
(N=17) |
Brigatinib
(N=7) |
1151Tins |
2% |
0% |
0% |
0% |
C1156Y |
2% |
8% |
0% |
0% |
I1171T/N/S |
2% |
4% |
12% |
0% |
F1174L/C |
0% |
17% |
0% |
0% |
V1180L |
0% |
4% |
6% |
0% |
L1196M |
7% |
8% |
6% |
0% |
G1202R |
2% |
21% |
29% |
43% |
G1202del |
0% |
8% |
0% |
0% |
D1203N |
0% |
4% |
0% |
14% |
S1206Y/C |
2% |
0% |
0% |
14% |
E1210K |
2% |
0% |
0% |
29% |
G1269A |
4% |
0% |
0% |
0% |
ALK Mutations |
20% |
54% |
53% |
71% |
Clinical strategies
Based on the results of clinical trials and FDA approval, alectinib is the preferred therapy for untreated patients with ALK-positive metastatic NSCLC. Crizotinib and ceritinib are also recommended in first-line setting [3]. In the phase III trial comparing crizotinib with chemotherapy (pemetrexed or docetaxel) as second line treatment, patients who continued crizotinb beyond disease progression showed a median duration of further treatment of 16 weeks (range 3-73 weeks) [5]. In a retrospective analysis of two single-arm trials, patients with disease progression who still obtain clinical benefits from crizotinib and who were allowed to continue treatment, showed a significantly longer overall survival than those who stopped the drug (16.4 vs. 3.9 months) [36]. Based on this results patients with asymptomatic progression can continue crizotinb. Patients experiencing “oligo-progressive disease” (progression in a single site or in up to five sites) can continue crizotinib in association with local therapy (surgery and/or ablative treatments) [3]. The availability of second- and third-generation ALK-inhibitors has extended the therapeutic options both for patients experiencing disease progression on crizotinib and for crizotinib-naive patients. Subsequent treatments for patients who progress on first-line crizotinib include alectinib or ceritinib or brigatinib. Similarly to crizotinib, patients with asymptomatic progression or oligoprogression on alectinib or ceritinib can continue the drug in association with local therapy. Cytotoxic therapy can be proposed to patients with systemic progression in multiple sites on crizotinib, alectinib or ceritinib [3] (Figures 3-5).
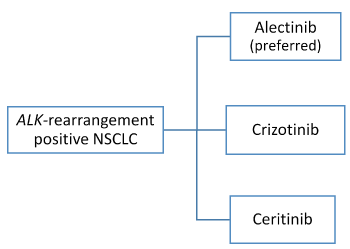
Figure 3. ALK-rearrangement positive NSCLC: first-line therapy [3]
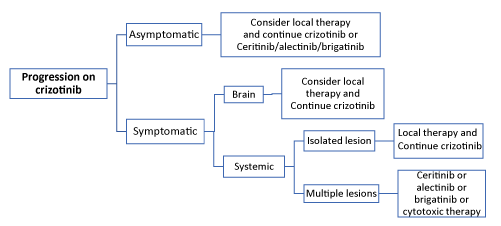
Figure 4. ALK-rearrangement positive NSCLC: subsequent therapy [3]

Figura 5. ALK-rearrangement positive NSCLC: subsequent therapy [3]
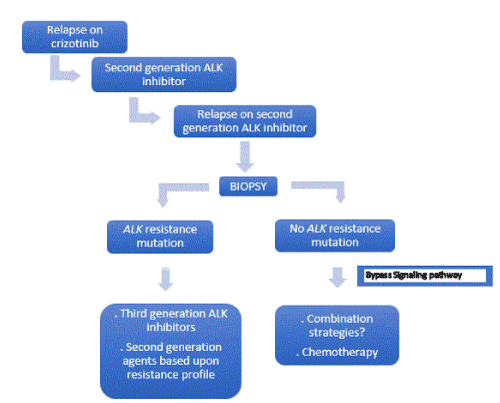
Figure 6. New paradigms and new opportunities of therapy [66]
Toxicity profile of ALK inhibitors
ALK-inhibitors demonstrated an overall interesting toxicity and safety profile. Regarding alectinib most adverse events were grade 1–2, the most frequent were constipation (36%), fatigue (33%), peripheral edema (25%) and myalgia (21%). Grade 3–4 events were mainly asymptomatic laboratory abnormalities: elevated gamma-GT, neutropenia, and hypophosphataemia [43,47]. The most common all-grade toxicity of ceritinib was diarrhea and nausea, reported in approximately 80% of patients. The most common grade 3–4 events were laboratory abnormalities: increased alanine and aspartate aminotransferase [38,40]. With regards to brigatinib in ALTA trial the most common any-grade adverse events included nausea (arm A/B, 33%/40%), diarrhea (arm A/B, 19%/38%), headache (arm A/B, 28%/27%). A subset of pulmonary adverse events (AE) caracterized by early onset (median time to onset 2 days, range 1 to 9 days), dyspnea, hypoxia, cough, pneumonia or pneumonitis occurred in 6% of patients. These AEs, occurring at 90 mg in boths arms without further events after escalation to 180 mg, requires early diagnoses and treatment [49]. As reguard to lorlatinib, in phase I clinical trial the most common treatment-related adverse events among the 54 enrolled patients were hypercholesterolaemia (72%), hypertriglyceridaemia (39%), peripheral neuropathy (39%), and peripheral oedema (39%). The most common grade 3-4 adverse event was hypercholesterolaemia. Neurologic and psychiatric disorders and peripheral neuropathy, occurring in 28% to 36% of patients, represent grade 1-2 toxicity but require rapid identification and management [60]. With regard to entrectinib, in the phase I study ALKA-372-001, the most common all-grade toxicity was paresthesia (42%), while asthenia represented the dose-limiting toxicity and muscular weakness the most common grade 3-4 events [62]. In the STARTRK-1 trial fatigue (33%) was the most common all-grade toxicity, neutropenia (11%), fatigue (7%) and cognitive impairment (7%) represented the most frequent grade 3–4 events [62].
Target agents and immune check-point inhibitors
Both in oncogene-addicted and molecularly unselected advanced NSCLC, mutational load impacts on tumor immunogenicity. Tumor cell death induced by chemotherapy and target agents produces the release of neoantigen triggering the immune response [66]. Clinical trials evaluating combination strategies with target agents in association with immune check-points inhibitors, including PD-1/PD-L1 and CTLA-4 inhibitors, are ongoing despite available data from CheckMate 057 [67] and KEYNOTE 010 [68] showed statistically significant shorter PFS and borderline lower ORR in EGFR-mutant/ALK-positive patients who are generally non-smokers [69] (lower mutational load and lower immunogenecity has been observed in never-smoker population). Early results from combination trial in selected TKI-naive or TKI-pretreated EGFR- or ALK-mutated NSCLC showed increased anti tumor responses although in some cases safety questions [70]. Enrollment in a Phase 1/2 study of nivolumab plus crizotinib in previously untreated ALK-positive NSCLC (CheckMate 370) was closed and combination treatment was discontinued due to observed grade ≥3 hepatic toxicities [71]. In a phase 1 dose escalation study in previously treated (ALK inhibitor or chemotherapy) or untreated IIIB/IV ALK-positive NSCLC, the combination of nivolumab plus ceritinib showed an interesting activity but a significant toxicities [72]. JAVELIN Lung 101 is a phase 1b/2 dose-finding trial evaluating avelumab plus crizotinib or aveumab plus lorlatinib in patients with advanced ALK-negative/wildtype NSCLC or ALK-positive NSCLC, respectively. The combination of avelumab and lorlatinib showed an acceptable safety profile, distinct from avelumab and crizotinib, and promising antitumor activity in patients with ALK-positive NSCLC. This combination will be evaluated in treatment-naive patients in phase 2 trial [73] (Table 2).
Table 2. Clinical trials of immune checkpoint inhibitors in combination with ALK TKIs in advanced NSCLC [72]
Clinical trial |
Phase |
Setting |
Intervention |
Status |
NCT02574078/
CheckMate 370 |
I/II |
Newly diagnosed/maintenance LA/
stage IV NSCLC |
Nivolumab+erlotinib (group D)/crizotinib (group E) |
Ongoing, not
recruiting for
group E |
NCT01998126 |
I |
Stages II–IV TKI-naïve or TKI-treated for less than 6 months EGFR- or ALKmutated NSCLC |
Nivolumab/ipilimumab+ erlotinib/crizotinib |
Ongoing, not
recruiting |
NCT02511184 |
I |
Newly diagnosed LA/stage IV ALKpositive non-squamous NSCLC |
Pembrolizumab +crizotinib |
Recruiting |
NCT02013219 |
I |
LA/stage IV TKI-naïve EGFR-mutated
and treatment-naïve ALK-positive
NSCLC |
Atezolizumab+erlotinib/
alectinib |
Ongoing, not
recruiting |
NCT02584634/
Javelin Lung 101 |
Ib/II |
LA/stage IV pretreated ALK-negative
(group A) or ALK-positive (group B)
NSCLC |
Avelumab+crizotinib (group A)/lorlatinib (group B) |
Recruiting |
NCT02898116 |
I/II |
Stage IV ALK rearranged NSCLC |
Durvalumab+ensartinib |
Recruiting |
NCT01998126 |
I |
Stages II–IV TKI-naïve or TKI-treated for less than 6 months EGFR- or ALKmutated NSCLC |
Ipilimumab+erlotinib/crizotinib |
Ongoing, not
recruiting |
Conclusions
Acquired resistance is the main concern of targeted-therapies in oncogene-addicted NSCLC. Even in ALK-rearranged NSCLC identification of mechanisms of acquired resistance, which can be target-dependent or target-independent, will be essential to design the best treatment strategy in each patient. Repeating biopsies after progression on TKIs will play a crucial role in treatment algorithm. Terapeutic strategies for non-target dependent acquired resistance still remain an open issue. New paradigms, including combination treatment and association with immunoterapy, are under investigation to identify new opportunities of therapy (Figure 6). The introduction of novel, more potent compound in untreated ALK-positive NSCLC highlights the need to develop studies evaluating sequences of targeted therapies versus upfront next-generation molecules to define which strategy could offer the longer control of disease.
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, et al. (2002) Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 346: 92-98. [Crossref]
- Cappuzzo F (2015) Guide to targeted therapies: Treatment resistance in lung cancer. Adis-Springer, Switzerland.
- NCCN guidelines (2018) Version 5. Last accessed June 27, 2018, www.nccn.org.
- Mino-Kenudson M, Chirieac LR, Law K, Hornick JL, Lindeman N, et al. (2010) A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res 16: 1561-1571.
- Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, et al. (2013) Crizotinib versus chemotherapy in advanced ALK-positive lung cancer.N Engl J Med368: 2385-2394. [Crossref]
- Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, et al. (1994) Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 263: 1281-1284. [Crossref]
- Yoshida T, Oya Y, Tanaka K, Shimizu J, Horio Y, et al. (2016) Differential crizotinib response duration among ALK fusion variants in ALK-positive non-small-cell lung cancer. J Clin Oncol 34: 3383-3389. [Crossref]
- Lindeman N, Cagle PT, Beasley MB, Chitale DA, Dacic S, et al. (2013) Molecular testing guidelines for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College og American Pathologist, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thoracic Oncol 8: 823-859. [Crossref]
- Camidge D, Kono SA, Flacco A, Tan AC, Doebele RC, et al. (2010) Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res 16: 5581-5590. [Crossref]
- Kwak El, Bang YI, Camidge DR, Shaw AT, Solomon B, et al. (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363: 1693-1703. [Crossref]
- Yoshida A, Tsuta K, Watanabe S, Sekine I, Fukayama M, et al. (2011) Frequent ALK rearrangement and TTF-1/p63 co-expression in lung adenocarcinoma with signet-ring cell component.Lung Cancer72: 309-315. [Crossref]
- Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, et al. (2012) Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study.Lancet Oncol13: 1011-1019. [Crossref]
- Kim DW, Ahn MU, Shi Y, De Pas T, Yang PC, et al. (2012) Results of a global phase II study with crizotinib in advanced ALK positive non-small cell lung cancer. J Clin Oncol 30: 7533.
- Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, et al. (2013) Crizotinib versus chemotherapy in advanced ALK-positive lung cancer.N Engl J Med368: 2385-2394. [Crossref]
- Mok T, Kim DW, Wu YL, Solomon BJ, Nakagawa K, et al. (2014) First-line crizotinib versus pemetrexed-cisplatin or pemetrexed-carboplatin in patients (pts) with advanced ALK-positive non-squamous non-small cell lung cancer (NSCLC): results of a phase III study (PROFILE 1014). J Clin Oncol 32: 8002.
- Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, et al. (2010) EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors.N Engl J Med363: 1734-1739. [Crossref]
- Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, et al. (2012) Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers.Sci Transl Med4: 120ra17. [Crossref]
- Costa DB, Shaw AT, Ou SH, Solomon BJ, Riely GJ, et al. (2015) Clinical experience with Crizotinib in patients with Advanced ALK-rearranged non- small-cell lung cancer and brain metastases. J Clin Oncol 33: 1881-1888. [Crossref]
- Solomon B, Felip E, Blackhall F, Mok TSK, Kim D, et al. (2014a) Overall and incranial (IC) efficacy results and time to symptom deterioration in PROFILE 1014: 1st line crizotinib vs. premetrexed-platinum chemotherapy (PPC) in patients with advanced ALK-positive non-squamous non-smeall cell lung cancer (NSCLC). Ann Oncol 25: iv426–iv470.
- Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, et al. (2014) First-line crizotinib versus chemotherapy in ALK-positive lung cancer.N Engl J Med371: 2167-2177. [Crossref]
- Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, et al. (2012) Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer.Clin Cancer Res18: 1472-1482. [Crossref]
- Sequist LV, Gettinger S, Senzer NN, Martins RG, Jänne PA, et al. (2010) Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer.J Clin Oncol28: 4953-4960. [Crossref]
- Chen Z, Sasaki T, Tan X, Carretero J, Shimamura T, et al. (2010) Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene.Cancer Res70: 9827-9836. [Crossref]
- Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, et al. (2013) Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov 3: 430-443. [Crossref]
- Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition.Nat Rev Mol Cell Biol15: 178-196. [Crossref]
- Gower A, Hsu WH, Hsu ST, Wang Y, Giaccone G (2016) EMT is associated with, but does not drive resistance to ALK inhibitors among EML4-ALK non-small cell lung cancer.Mol Oncol10: 601-609. [Crossref]
- Guo F, Liu X, Qing Q, Sang Y, Feng C, et al. (2015) EML4-ALK induces epithelial mesenchymal transition consistent with cancer stem cell properties in H1299 non-small cell lung cancer cells. Biochem Biophys Res Commun 459: 398-404. [Crossref]
- Kim HR, Kim WS, Choi YJ, Choi CM, Rho JK, et al. (2013) Epithelial-mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4-ALK translocation. Mol Oncol 7: 1093-1102. [Crossref]
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, et al. (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3: 75ra26. [Crossref]
- Cha YJ, Cho BC, Kim HR, Lee HJ, Shim HS (2016) A case of alk-rearranged adenocarcinoma with small cell carcinoma-like transformation and resistance to Crizotinib.J Thorac Oncol11: e55-55e58. [Crossref]
- Miyamoto S, Ikushima S, Ono R, Awano N, Kondo K, et al. (2016) Transformation to small-cell lung cancer as a mechanism of acquired resistance to crizotinib and alectinib.Jpn J Clin Oncol46: 170-173. [Crossref]
- Pop O, Pirvu A, Tof2021 Copyright OAT. All rights reserv flare after treatment discontinuation in a patient with EML4-ALK lung cancer and acquired resistance to crizotinib. J Thorac Oncol 7: e1–e2. [Crossref]
- Kuriyama Y, Kim Y, Nagai H, Ozasa H, Sakamori Y, et al. (2013) Disease flare after discontinuation of crizotinib in anaplastic lymphoma kinase-positive lung cancer. Case Rep Oncol 6: 430–433. [Crossref]
- Weickhardt A, Scheier B, Burke J, Gan G, Lu X, et al. (2012) Local ablative therapy of oligoprogressive disease prolongs disease control by tyrosine kinase inhibitors in oncogeneaddicted non-small-cell lung cancer. J Thorac Oncol 7: 1807–1814. [Crossref]
- Romanidou O, Landi L, Cappuzzo F, Califano R (2016) Overcoming resistance to first/second generation epidermal growth factor receptor tyrosine kinase inhibitors and ALK inhibitors in oncogene-addicted advanced non-small cell lung cancer. Ther Adv Med Oncol 8: 176-187. [Crossref]
- Ou SH, Jänne P, Bartlett C, Tang Y, Kim DW, et al. (2014) Clinical benefit of continuing ALK inhibition with crizotinib beyond initial disease progression in patients with advanced ALK-positive NSCLC. Ann Oncol 25: 415-422. [Crossref]
- Lim SM, Kim HR, Lee JS, Lee KH, Lee YG, et al. (2017) Open-Label, multicenter, phase ii study of ceritinib in patients with non-small-cell lung cancer harboring ROS1 rearrangement.J Clin Oncol35: 2613-2618. [Crossref]
- Kim DW, Mehra R, Tan DSW, Felip E, Chow LQM, et al. (2016) Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial.Lancet Oncol17: 452-463. [Crossref]
- Mok T, Spigel D, Felip E, deMarinis F, Ahn MJ, et al. (2015) ASCEND-2: A single arm, open-label, multicenter phase 2 study of ceritinib in adult patients (pts) with ALK-rearranged (ALK+) non-small cell lung cancer (NSCLC) previously treated with chemotherapy and crizotinib (crz). J Clin Oncol 33: 8059.
- Felip E, Orlov S, Park K, Yu CJ, Tsai CM, et al. (2015) ASCEND-3: A single-arm,open-label, multicenter phase 2 study of ceritinib in ALK naive adult patients (pts) with ALK-rearranged (ALK+) non-small cell lung cancer (NSCLC). J Clin Oncol 33: 8060.
- Soria JC, Tan DSW, Chiari R, Wu YL, Paz-Ares L, et al. (2017) First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 389: 917-929. [Crossref]
- Shaw AT, Kim TM, Crinò L, Gridelli C, Kiura K, et al. (2017) Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 18: 874-886. [Crossref]
- Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, et al. (2017) Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer.N Engl J Med377: 829-838. [Crossref]
- Hida T, Nokihara H, Kondo M, Kim YH, Azuma K, et al. (2017) Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): an open-label, randomised phase 3 trial.Lancet390: 29-39. [Crossref]
- Larkins E, Blumenthal GM, Chen H, He K, Agarwal R, et al. (2016) FDA approval: Alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following Crizotinib.Clin Cancer Res22: 5171-5176. [Crossref]
- Shaw AT, Gandhi L, Gadgeel S, Riely GJ, Cetnar J, et al. (2016) Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. Lancet Oncol 17: 234-242.
- Ou SH, Ahn JS, De Petris L, Govindan R, Yang JC, et al. (2016) Alectinib in Crizotinib-refractory ALK-rearranged non-small-cell lung cancer: A phase II global study.J Clin Oncol34: 661-668. [Crossref]
- Zhang S, Anjum R, Squillace R, Nadworny S, Zhou T, et al. (2016) The potent ALK inhibitor Brigatinib (AP26113) overcomes mechanisms of resistance to first- and second-generation ALK inhibitors in preclinical models. Clin Cancer Res 22: 5527-5538. [Crossref]
- Kim DW, Tiseo M, Ahn MJ, Reckamp KL, Hansen KH, et al. (2017) Brigatinib in patients with Crizotinib-refractory anaplastic lymphoma kinase–positive non–small-cell lung cancer: A randomized, multicenter phase ii trial. J Clin Oncol 35: 2490-2498. [Crossref]
- Tiseo M, Popat S, Gettinger SN, Peters S, Haney J, et al. (2018) Design of ALTA-1L (ALK in lung cancer trial of brigatinib in first-line), a randomized phase 3 trial of brigatinib (BRG) versus crizotinib (CRZ) in tyrosine kinase inhibitor (TKI)-naive patients (pts) with advanced anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC). J Clin Oncol 35.
- Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, et al. (2015) PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to 1st and 2nd generation ALK inhibitors in pre-clinical models. Cancer Cell 28: 70-81. [Crossref]
- Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, et al. (2012) Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer.Clin Cancer Res18: 1472-1482. [Crossref]
- Kim S, Kim TM, Kim DW, Go H, Keam B, et al. (2013) Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J Thorac Oncol 8: 415–422. [Crossref]
- Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, et al. (2012) Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers.Sci Transl Med4: 120ra17. [Crossref]
- Shaw AT, Engelman JA (2013) ALK in lung cancer: past, present, and future.J Clin Oncol31: 1105-1111. [Crossref]
- Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, et al. (2014) Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem 57: 4720–4744. [Crossref]
- Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, et al. (2014) Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dosefinding portion of a phase 1/2 study. Lancet Oncol 15: 1119–1128. [Crossref]
- Shaw AT, Mehra R, Tan D, Felip E, Chow LQ, et al. (2014b) 1293P - Evaluation of ceritinib-treated patients (pts) with anaplastic lymphoma kinase rearranged (ALK+) non-small cell lung cancer (NSCLC) and brain metastasis. Ann Oncol 25: iv426.
- Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, et al. (2016) Resensitization to Crizotinib by the Lorlatinib ALK resistance mutation L1198F.N Engl J Med374: 54-61. [Crossref]
- Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, et al. (2017) Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial.Lancet Oncol18: 1590-1599. [Crossref]
- ClinicalTrials.gov, A study of Lorlatinib versus Crizotinib in first line treatment of patients with ALK-positive NSCLC. Identifier: NCT03052608.
- Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, et al. (2017) Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor Entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov 7: 400–409. [Crossref]
- Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, et al. (2017) Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1).Cancer Discov7: 400-409. [Crossref]
- Rangaraju S, Li G, Christiansen J, Hornby Z, Multani P, et al. (2017) TRTH-10. Pediatric Phase 1/1B Study of entrectinib in patients with primary brain tumors, neuroblastoma, and NTRK, ROS1, or ALK Fusions. Neuro-Oncology 19: iv53.
- Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, et al. (2016) Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer.Cancer Discov6: 1118-1133. [Crossref]
- Pilotto S, Molina-Vila M, Karachaliou N, Carbognin L, Viteri S, et al. (2015) Integrating the molecular background of targeted therapy and immunotherapy in lung cancer: a way to explore the impact of mutational landscape on tumor immunogenicity. Transl Lung Cancer Res 4: 721–727. [Crossref]
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, et al. (2015) Nivolumab versus Docetaxel in advanced nonsquamous non-small-cell lung cancer.N Engl J Med373: 1627-1639. [Crossref]
- Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, et al. (2016) Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387: 1540-1550. [Crossref]
- Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, et al. (2016) EGFR Mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: A retrospective analysis.Clin Cancer Res22: 4585-4593. [Crossref]
- Moya-Horno I, Viteri S, Karachaliou N, Rosell R (2018) Combination of immunotherapy with targeted therapies in advanced non-small cell lung cancer (NSCLC).Ther Adv Med Oncol10: 1758834017745012. [Crossref]
- Spigel DR, Reynolds C, Waterhouse D, Garon EB, Chandler J, et al. (2018) Phase 1/2 study of the safety and tolerability of Nivolumab plus Crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation — positive advanced non–small cell lung cancer (CheckMate 370). J Thorac Oncol 13: 682–688. [Crossref]
- Felip E, De Braud FG, Maur M, Loong HHF, Shaw AT, et al. (2017) Ceritinib plus nivolumab (NIVO) in patients (pts) with anaplastic lymphoma kinase positive (ALK+) advanced non-small cell lung cancer (NSCLC). J Clin Oncol 35: 2502-2502.
- AT Shaw, Lee SH, Ramalingam SS, Bauer TM, Boyer MJ et al. (2018) Avelumab (anti–PD-L1) in combination with crizotinib or lorlatinib in patients with previously treated advanced NSCLC: Phase 1b results from JAVELIN Lung 101. J Clin Oncol 36: abstr 9008.
- Camidge DR, Kim HR, Ahn MJ, Yang JCH, Han JY, et al. (2018) Brigatinib versus crizotinib in ALK-positive Non-Small-Cell Lung Cancer. N Engl J Med.