Acute myeloid leukemia is the primary acute leukemia affecting adults and until recently had very limited available treatment options. We are now seeing development and redevelopment of targeted therapy in acute myeloid leukemia with subsequent improvements in disease control and overall survival. The development of therapy with molecular targets allows patients who are not candidates for chemotherapy to receive treatment. In this review, we discuss the molecular mutations FLT3 and IDH and the molecular target CD33, recognized for having key roles in the development of acute myeloid leukemia. We reference key clinical trials to further discuss the approved treatments for acute myeloid leukemia that target these molecular mutations. We then discuss ongoing research to develop new drugs and new combinations of therapy to overcome resistance mechanisms and further improve patient outcomes.
acute myeloid leukemia, CD33, FLT3, IDH1, IDH2, molecular mutations, targeted therapy
Acute myeloid leukemia (AML) is an aggressive malignancy resulting from driving molecular mutations and chromosomal abnormalities [1]. Despite extensive efforts to understand the biology and mechanisms of leukemogenesis, the backbone of therapy has not changed since the 1970s, since introduction of cytarabine and anthracycline-based intensive induction chemotherapy (often referred to as “7+3”). Advances in survival have been largely driven by improved risk-stratification and treating eligible high-risk patients with allogeneic stem cell transplant [2,3]. Risk stratification takes into account cytogenetic (karyotype) and molecular categories of prognosis; usually defined as good, intermediate, or poor risk [2].
AML is primarily a disease of older adult patients, median age at diagnosis is 68 [4]. Many patients are not sufficiently fit to receive intensive induction chemotherapy followed by consolidation chemotherapy and possibly maintenance treatment or allogeneic stem cell transplant. Some patients will have comorbidities (such as congestive heart failure) that make anthracycline-based therapy relatively contraindicated. While lower intensity options, such as hypomethylating agents and low dose cytarabine may offer some benefit, there remains a large unmet need for many AML patients.
Between 2017 and 2019, the Food and Drug Administration (FDA) approved eight new therapeutic agents for treatment of AML. Many of these agents utilize unique molecular targets and provide treatment options to patients who historically had a limited therapeutic profile. We present a summary of these agents, their biological principles, and their utilization in AML. Additionally, patient cases are included as a testament to the efficacy of these new agents and their potential to change both the therapeutic opportunities and prognosis of patients with AML.
Molecular mutations
FMS-like tyrosine kinase 3 (FLT3) is a class III receptor tyrosine kinase, a proto-oncogene and is important in hematopoiesis as it is present on hematopoietic stem cells and helps regulate cell proliferation and apoptosis [5]. There are 2 common mutations of FLT3, internal tandem duplications (ITD) and tyrosine kinase domain (TKD) point mutations. These are the most common genetic mutations in AML, with ITDs seen in up to 35% and TKDs in up to 10% of patients. FLT3 Internal tandem duplications (FLT3-ITD) are usually within exon 14, though occasionally exon 15 is involved. This area usually encodes for the juxtamembrane domain (JM) of the receptor, see Figure 1 [6]. The ITD mutation promotes ligand-independent autophosphorylation and constitutive activation allowing AML cells to proliferate and survive [7]. The presence of this mutation has been historically associated with decreased overall survival (OS) with a median OS of 6-12 months [8,9]. Tyrosine kinase domain point mutations, most often in codon D835 or deletion of codon I836, lead to a gain of function that allows ligand-independent receptor autophosphorylation [10]. There is some evidence presence of this mutation is slightly more favorable than FLT3-ITD, especially with a higher mutation burden [11]. These mutations were initially discovered in the early to mid-1990s and over the years, more information on these prognostic markers becomes available. Due to prevalence of these mutations, there has been much research over the past 20 years looking into therapies directed at these mutations.
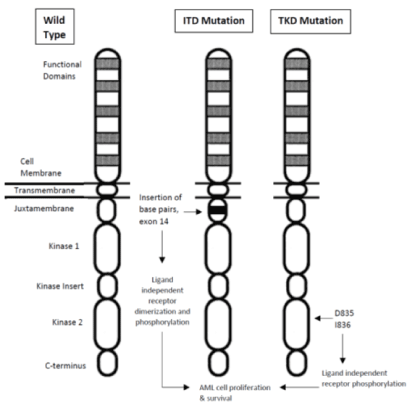
Figure 1. FLT3 Mutations
Treatment strategies in FLT3-mutated AML
Initial treatment strategies in FLT3-mutated AML begin with 1) evaluating the patient and their performance status, 2) age of the patient and 3) cytogenetics and molecular studies. Patients with FLT3-mutated AML are risk stratified based on their overall cytogenetic and molecular mutations. Most patients with FLT3-mutated AML fall into either intermediate or poor-risk prognostic stratification [3]. Because of these risks, there is ongoing emphasis on research into FLT3 directed therapy. Targeted agents (small molecule inhibitors) directed at FLT3 mutations have been approved in recent years, and more recently, have been incorporated into induction, consolidation and maintenance chemotherapy regimens [12-14].
Midostaurin (Rydapt®) was FDA approved in 2017, after results of the Cancer and Leukemia Group B (CALGB) research cooperative RATIFY trial showed a statistically significant increase in overall survival (OS) and event free survival (EFS) with the addition of midostaurin to induction chemotherapy compared with standard of care chemotherapy, in FLT3-mutated AML patients [12-14]. The 4-year OS for the midostaurin group was 51.4% compared the 44.7% in the placebo group (p = 0.009) [14]. AML patients are placed into intensive therapy and non-intensive therapy candidate categories. Historically, patients under 60 years of age or younger are considered induction (intensive therapy) candidates with high doses of chemotherapy. Eligible patients received induction chemotherapy with the addition of midostaurin (50 mg) orally every 12 hours on days 8 through 21 of a treatment cycle. If residual disease is present at day 21, an additional cycle of induction chemotherapy with midostaurin can be given. If a complete response (CR) is obtained, consolidation chemotherapy is given with midostaurin for 2-3 treatments [15]. Ultimately the treatment goal is to get patients to an allogeneic stem cell transplant which is considered curative.
While the hopes of either obtaining CR or cure after allogeneic stem cell transplant is the goal, and occurs in some patients, many will relapse or be refractory to induction chemotherapy indicating resistance to treatment. In 2018, gilteritinib (Xospata®)was approved for use in relapsed/refractory FLT3-mutated AML after results from the ADMIRAL trial showed the oral, once daily, single agent, small molecular inhibitor, had higher incidences of remission than salvage chemotherapy [16,17]. See Case 1 for a real patient example. Because of the mortality associated in patients who are deemed unsuitable for induction chemotherapy, alternative strategies are used to safely treat this patient population. Standard of care combines non-intensive hypomethylating agents azacitidine or decitabine with sorafenib as this combination is well tolerated with good response rates, including a low percentage of complete responders [18].
Development of resistance to FLT3 inhibitors is a treatment challenge and provides rationale for research in combining FLT3 inhibitors with conventional and investigational therapy [19]. There is currently an ongoing clinical trial comparing induction and consolidation chemotherapy (NCT03836209) with midostaurin vs gilteritinib [20]. Another clinical trial with the FLT3 inhibitor quizartinib, combined with vyxeos induction chemotherapy is ongoing as well, though neither agent has approval in relapsed/refractory AML by itself [21,22]. Additionally crenolanib is being studied in the same setting, though did not gain approval in its clinical trial and is being currently studied in the maintenance phase of AML treatment [23,24].
Molecular mutations
Isocitrate dehydrogenase (IDH) mutations in AML impede conversion of isocitrate to alpha ketogluconate (a-KG) in the Krebs cycle. Instead, IDH mutations generate 2-hydroxyl glutarate (2-HG), an ‘onco-metabolite’ which interferes with cellular regulatory mechanisms, ultimately halting cell differentiation and promoting leukemic tumor genesis [25-27]. IDH exists in three different iso-forms, IDH1, IDH2 and IDH3. Mutations in IDH1 and IDH2 have been identified in multiple cancers; glioma, cholangiocarcinoma, chondrosarcoma, AML and others [28]. IDH1 and IDH2 mutations are found in approximately 15-20% of AML cases and are more frequently identified in older patients and those with intermediate risk karyotype [29]. Their prognostic impact is unclear due to conflicting data. IDH2 subtype mutations such as IDH2 R140Q and IDH2 R172 have been associated with a better prognosis but co-occurrence with FLT3 mutations appear to nullify this benefit [30-32]. The presence of IDH mutations are mutually exclusive and they frequently accompany NPM1 and FLT3 mutations. They have also been found to be mutually exclusive with TET2 mutations [29,33]. IDH mutations are heterogeneous, different sub-types have prognostic implications and some such as IDH2 R172 have a poor response to conventional AML directed chemotherapy [30]. Hence there is a role for specific treatment which targets IDH mutations in AML. The Food and Drug administration (FDA) has approved two agents, Ivosidenib (Tibvosoâ) and Enasidenib (Idhifaâ) for treatment of adults with IDH1 and IDH2 mutated AML [34,35].
Treatment strategies in IDH mutated AML
Ivosidenib, (TIBVOSOâ), an IDH1 Inhibitor was approved by the FDA in July 2017 for treatment of adults with relapsed/refractory AML with IDH1 mutations after a phase I/II study of 179 patients with IDH1 mutated relapsed/refractory AML treated with orally administered Ivosidenib daily reported a 42% overall response rate (ORR), including 30% complete response (CR). The median duration of response was greater than 8 months. Over a third of patients attained transfusion independence which is a meaningful quality of life outcome [35]. See Case 2 for a real patient example. In May 2019, Ivosidenib received FDA approval for treatment of newly diagnosed AML with an IDH1 mutation in adults 75 years or older, or in adults unable to get intensive chemotherapy due to co-morbidities. The approval was based on a phase I study of 34 patients (median age 76) with IDH1 mutated AML which found a median OS of 13 months, median follow up in the study was 2 years. The expected survival of patients with newly diagnosed AML who cannot receive intensive chemotherapy is about 6 months. The single agent Ivosidenib achieved 30% CR, in addition 9 of 21 transfusion-dependent patients became transfusion-independent [36]. Ivosidenib carries a black box warning for differentiation syndrome. Due to rapid cancer cell maturity and death, differentiation syndrome is a clinical syndrome of dyspnea, fever, weight gain, hypertension, renal failure, and pulmonary infiltrates on chest radiograph [37]. Other toxicities associated with Ivosidenib treatment are diarrhea, febrile neutropenia, nausea, fatigue and prolonged QTc interval. Other inhibitors of IDH1 mutated proteins aimed at inhibiting 2-HG levels and tumor proliferation are under investigation in early phase trials, either as monotherapies or in combination with hypomethylating agents and traditional chemotherapy.
Enasidenib, (IDHIFA®), an IDH2 inhibitor is an oral agent approved by the FDA August 2017 for treatment of IDH2 mutated, relapsed/refractory AML. Enasidenib was shown to reduce levels of the ‘oncometabolite’ 2-HG by greater than 90% and induce differentiation of AML cells [38]. A study of 214 patients treated with Enasidenib 100mg daily found that 19% achieved CR with a median OS of 9 months [39]. Treatment related toxicities were elevated bilirubin, nausea, diarrhea and 14% of patients experienced grade ≥ 3 toxicities. Differentiation syndrome occurred in 14% of patients, and half of those experienced grade 3 or higher differentiation syndrome treated with steroids, hydroxyurea and diuretics [39]. Resistance has been documented to Ivosidenib and Enasidenib, the mechanism is unclear but alterations in NRAS and MAPK pathways have been implicated [39]. IDH inhibitors in combination with hypomethylating agents and conventional chemotherapy are being studied in clinical trials to address resistance.
Venetoclax (Venclexta®), an indirect IDH inhibitor, is an oral, highly selective inhibitor of the B-cell lymphoma 2 (BCL-2) receptor. BCL-2 is an anti-apoptotic gene which works with mutated IDH to promote leukemic cell survival [40]. A clinical trial of single agent Venetoclax in relapsed/refractory IDH mutated AML found a combined CR and complete response with incomplete count recovery (Cri) rate of 33% indicating the promise of venetoclax in IDH mutated AML [41].
Cell surface marker
CD33 is a transmembrane receptor expressed by malignant cells of myeloid lineage [42]. The extracellular part of the receptor contains immunoglobulin domains IgV and IgC2 while the intracellular portion contains immunoreceptor tyrosine-based inhibitory motifs. CD33 is stimulated by any glycoproteins or glycolipids with sialic acid residues and the resultant cascade of actions after binding inhibits cellular phagocytosis [43]. CD33 is not found on normal mature hematopoietic cells, making it an ideal target for AML therapy [42].
Treatment strategies in CD33 positive AML
Gemtuzumab ozogamicin (Mylotarg®) was the first monoclonal antibody for AML, as well as one of the only medications to ever gain FDA approval twice for the same indication, first in 2000 and again in 2017. It was initially granted accelerated approval for patients age 60 or greater with CD33+ relapsed AML [44].
Gemtuzumab ozogamicin was granted accelerated approve based on three phase II studies that demonstrated CR or CRi in 30% of adult patients, with the requirement phase III studies be completed to show benefit over current therapy. Ultimately the phase III Southwest Oncology Group (SWOG) S0106 trial found no benefit of gemtuzumab ozogamicin plus chemotherapy versus chemotherapy alone, and actually demonstrated a sizeable increase in treatment related deaths [45]. The most common causes of treatment related deaths were infection, acute respiratory distress syndrome and hemorrhage. This data combined with two additional phase III studies with negative results, led to voluntary withdrawal of gemtuzumab ozogamicin from the market in 2010 [46]. The FDA reapproved gemtuzumab ozogamicin on September 1, 2017 for both newly diagnosed and relapsed/refractory CD33-postitive AML, with decreases in dosing leading to improved safety and efficacy [47]. The pivotal trial that led to reapproval was the French multicenter study, ALFA-0701, with an endpoint of 3 year event free survival of 39.8% for gemutuzumab ozogamicin and chemotherapy compared to 13.6% for chemotherapy alone (p<0.001) [48]. At the time of re-approval, the 5 year OS for AML was 27.4%, while a substantial jump from 6.3% in 1975 when 7+3 became standard of care, there still remained significant area for improvement.
Gemtuzumab ozogamicin is an intravenous, humanized, anti-CD 33 monoclonal antibody linked to a derivative of a potent antibiotic, calicheamicin, which is rapidly internalized after binding. In AML the white blood cells do not mature, leaving many CD33 receptors on the surface of AML blasts. Multiple studies have shown CD-33 positive cells are quick to internalize antibodies after binding (Figure 2) [49]. The peptide that links the antibody to calicheamicin was designed to only release within the acidic environment of the blast, ultimately binding to the DNA, leading to double strand breaks and cell death [50].
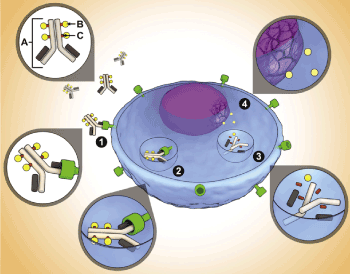
Figure 2. Antibody Drug Conjugate (ADC) structure and mechanism of action
(A): CD-33 monoclonal antibody; (B) calicheamicin; (C) functional linker that binds calicheamicin to CD-33 antibody. ADC mechanism of action: (1) binding of ADC to CD-33 on blast; (2) internalization of antigen-ADC complex; (3) lysosomal degradation releases calicheamicin from antibody; (4) drug causes DNA double strand bread and ultimately death. Permission for re-print pending
It should be noted gemtuzumab ozogamicin causes significant liver toxicity, including possibly fatal veno-occlusive disease (VOD). In ALFA-0701, VOD was reported in 5% of patients who received gemtuzumab ozogamicin prior to or following allogeneic stem cell transplant [48]. Gemtuzumab ozogamicin is not considered an intensive therapy, making it ideal for single agent treatment for older or frail patients. It can also be used in combination with intensive chemotherapy in patients with favorable or intermediate risk disease [for example, core binding factor or normal cytogenetics]. Given potentially fatal liver damage, there is a black box warning on the medication for patients who have received or plan to receive a stem cell transplant [3].
Studies are ongoing to determine gemtuzumab ozogamicin’s role in different patient populations and subtypes of AML. Acute Promyelocytic leukemia (APL) makes up 10-12% of AML but has a high surface expression of CD33. Single agent gemtuzumab ozogamicin has shown durable remission rates in relapsed APL [51].
Future directions to target molecular mutations in AML
Further research is needed to identify new targets for directed treatments in AML. The Eastern Cooperative Oncology Group (ECOG) analyzed somatic mutations in 398 AML patients, finding 97.3% had at least 1 identifiable mutation among 18 genes tested [31]. All of these mutations are involved in AML development and communicate by downstream effects, adaptive feedback, and crosstalk underscoring the complexity of AML and its ability to develop resistance to targeted therapy [52]. The number of novel therapeutic agents for existing and new targets is expanding exponentially. A search on clinicaltrials.gov yields over 2000 results, even limiting to phase 1 and phase 2 studies. Restricting the search to studies actively recruiting adult AML patients still yields an impressive 419 studies, see Table 1. The Cancer Genome Atlas Network whole exome and whole genome sequencing performed on 200 de novo AML samples demonstrated AML is characterized by few mutations in coding genes, averaging 13 per patient, 5 of these recurrent mutations, which is much less than solid tumors. These include FLT3 ITD, IDH1, IDH2 in addition to NPM1, DNMT3A, and TP53. FLT3 ITD, IDH1, and IDH 2 pathways and therapeutic interventions have been discussed. Other targets are being discovered as well.
Table 1. AML studies actively recruiting, clinicaltrials.gov
Terms |
Search Results* |
Entire Database** |
| Synonyms
|
AML |
419 studies |
2,847 studies |
Acute myeloid leukemia |
362 studies |
2,377 studies |
Acute myelogenous leukemia |
53 studies |
452 studies |
Acute myelocytic leukemia |
1 study |
18 studies |
Acute Myeloblastic Leukemias |
1 study |
202 studies |
Acute Nonlymphocytic Leukemia |
1 study |
7 studies |
Leukaemias acute myeloid |
1 study |
3 studies |
Acute granulocytic leukemia |
-- |
1 study |
Acute monoblastic leukemia |
-- |
179 studies |
Acute monocytic leukemia |
-- |
182 studies |
Acute non lymphoblastic leukemia |
-- |
2 studies |
BCL2 inhibitors
Evading apoptosis is a hallmark of cancer and hypothesized to cause resistance to chemotherapy. B-cell leukemia/lymphoma-2 (BCL-2) is an anti-apoptotic protein that promotes leukemic blast survival by regulating the mitochondrial apoptotic pathway. Sensitizer BCL-2 homology 3 (BH3) proteins are antagonists of these antiapoptotic proteins and thus promote apoptosis via mitochondrial outer membrane permeabilization [53]. Venetoclax is a highly selective, oral small molecule inhibitor of BCL2. Venetoclax was studied at daily dose of 800mg in a phase 2, single are study of patients with high-risk relapsed/refractory AML or unfit for intensive chemotherapy. The overall response rate was 19%; an additional 19% of patients demonstrated anti-leukemic activity not meeting International Working Group response criteria (partial bone marrow response and incomplete hematologic recovery). Patients with IDH 1 and IDH2 mutations had higher response rates [53].
In a phase 1b, non-randomized, open label dose escalation study, previously untreated patient age 65 year or older ineligible for intensive chemotherapy were enrolled. Patient were required to have an Eastern Cooperative Group performance status of 0-2 and either intermediate or poor-risk cytogenetics. Overall, 61% of patients achieved CR or Cri [54]. Another large, multicenter, phase 1b escalation and expansion study evaluated venetoclax with hypomethylating agents (HMA). Patients were 65 years or older with treatment naïve AML and ineligible for intensive chemotherapy. Median age was 74 years, with poor risk cytogenetics in 49% of patients. With a median time on study of 8.9 months, 67% of patients achieved CR or CRi, with a CR/CRi rate of 73% in the venetoclax 400 mg and HMA cohort. The median DOR was 11.3 months and median OS 17.5 months [55]. Venetoclax with non-intensive chemotherapy cytarabine in phase I/II study demonstrated CR/CRi rates of 35% for secondary AML and 71% for de novo AML [56].
In a double-blind placebo-controlled phase 3 study (NCT03069352) 211 patients were randomized to venetoclax or placebo plus low dose cytarabine. With a median follow-up of 17.5 months, median OS was 8.4 months vs 4.1 months in the Venetoclax+cytarabine and placebo+cytarabine arms (HR 0.70; 95% CI 0.50–0.99; P=.04), representing a 30% reduction in risk of death. CR/CRi and CR/CRh (CR with partial hematologic recovery) rates were both 48% for the Venetoclax+cytarabine arm, and 13% and 15%, respectively, for placebo+cytarabine [57]. In a phase III randomized double-blinded multi-center trial (VIALE-A), efficacy of the combination of azacitidine and venetoclax in treatment-naïve AML patients ineligible for intensive therapy was compared to treatment with azacytidine plus a placebo. In 431 internationally enrolled patients in the VIALE-A trial, combination of azacitidine and venetoclax led to improved OS (14.7 vs 9.6 months), and improved CR/CRi (66% vs 28%), compared to azacitidine alone. In addition, the combination was associated with responses occuring more quickly (median time to CR/CRi was only 1.3 months) and more durable (lasting 1.5 years), with increased incidence of transfusion independence (58% vs 34%) [58].
Venetoclax is being studied in combination with IDH1 targeted agent Ivosidenib with/without azacitidine.(clinicaltrials.gov identifier: NCT 03471260) A recent study reported 19 patients with IDH1 mutation (median age 68),17 with AML: 9 relapsed/refractory AML (R/R; median 1 prior line of therapy), 5 treatment naïve AML, and 3 HMA-failure myelodysplastic syndrome (MDS) with secondary AML. In evaluable patients (n=18), composite complete remission (CRc: CR+CRi+CRh) rates were 78% overall (treatment naive: 100%, R/R: 75%), (median time to best response: 2 months) [59].
Myeloid cell leukemia inhibitors
MCL-1, like BCL-2, can block pro-apoptotic proteins like BAX and BAK. MCL 1 inhibitor was found to synergistically induce apoptosis with venetoclax in AML cell lines and primary patient samples [60]. MCL-1 inhibitors have been tested in early phase trials. However, trials were put on hold due to concerns of cardiotoxicity.
Targeting p 53
The tumor suppressor protein p53, encoded by TP53 gene in humans, plays a vital role in preventing oncogenesis. Mutation of the tumor suppressor gene tumor protein 53 (TP53) is found in over 50% of human cancers [61]. A mutation leads to protein unfolding, preventing this transcription factor from responding to cellular stress to elicit cell cycle arrest and apoptosis allowing for uncontrolled proliferation of cancer cells [62]. The incidence of TP53 mutations in AML is variable. It is reported in about 10% of de novo AML, 30% of AML after prior chemotherapy, and 70% of elderly patients with complex-karyotype AML. The TP53 mutation carries the worst prognosis AML conferring strong resistance to chemotherapy and allogeneic stem cell transplant [63]. APR-246 is a novel TP53-activating drug being studied in phase 1b/II clinical trials in combination with azacitidine in AML with report of CR rates of 60% and overall response rates greater than 70% [64,65]. APR-246 is now being studied in combination with venetoclax and azacitidine and in combination with azacitidine following allogeneic stem cell transplant for TP53 mutated AML [66,67]. In the phase 2 GFM-APR trial, treatment with eprenetapopt (APR-246) in combination with azacytidine achieved high objective response (ORRs) and CR rates in patients with TP53-mutated MDS and AML. Patients with AML who had 20% to 30% blasts had an ORR and CR rate of 78% and 33%, respectively [68].
Mouse double minute 2(Mdm2) inhibitor
Mdm 2, also known as E3 ubiquitin-protein ligase is a negative regulator of the p53 tumor suppressor. Mdm 2 binds to p53 and induces its proteasomal degradation. Mdm 2 is frequently overexpressed in AML retaining wild-type TP53 alleles. Several Mdm2 targeting agents are in clinical trials. A phase 1b study of evaluated MDM2 Inhibitor AMG 232 with or without trametinib in relapsed/refractory AML. Of 30 evaluable patients, 1 achieved CR, 4 had morphologic leukemia-free state, and 1 had partial remission. Four of 13 (31%) TP53-wild-type patients and 0 of 3 (0%) TP53-mutant patients were responders [69]. Idasanutlin is a small-molecule inhibitor of MDM2 and is currently being evaluated in a randomized phase III trial in combination with cytarabine in relapsed /refractory AML. The primary end point is OS in the TP53-WT population. (clinicaltrials.gov identifier: NCT 02545283).
Microenvironment targeting agents
AML blasts express receptors on their surface, causing interactions with the marrow microenvironment. These interactions have been implicated in both chemotherapy resistance and disease relapse and targeting the tumor-microenvironment interactions in AML is rational. Agents in this space include hypoxia-inducible agents and inhibitors of CXCR4 and adhesion molecules such as VLA-4 and E-selectin. E-selectin is a cell adhesion molecule involved in migration of leukocytes along vascular endothelial cells and is highly expressed on leukemic blasts, especially those with advanced disease [70]. Uproleselan (GMI-1271) is an antagonist of E-selectin which enhances chemotherapy response while ameliorating chemotherapy toxicity in animal models. In the phase I/II trial combining Uproleselan with Mitoxantrone, Etoposide, and Cytarabine (MEC) intensive chemotherapy in relapsed/refractory patients, the CR/CRi rate was 41%, with a 30- and 90-day mortality of 2% and 9% respectively [71]. In an elderly high-risk treatment-naïve AML population treated with Uproleselan combined with standard induction; 68% (73% de novo, 64% sAML) achieved CR/CRi. The 30- and 90-day mortality was 8% and 12%, respectively [72]. A phase III randomized trial is ongoing for relapsed/refractorty patients comparing Uproleselan with MEC versus MEC alone (ClinicalTrials.gov identifier: NCT03616470), and another phase II–III trial evaluating Uproleselan with induction versus induction alone are underway.
Hedgehog pathway inhibition
Normal vertebrate embryogenesis is controlled by hedgehog pathway (Hh), controlled by transmembrane proteins Patched (PTCH) and Smoothened (SMO). Hedgehog pathway controls normal growth and differentiation of adult tissues [73]. Aberrant signaling in hedgehog pathway affects proliferation of leukemia stem cells and upregulation of this pathway has been implicated in chemoresistance seen in AML [74]. Glasdegib is an oral agent inhibiting the Hh pathway by interacting with smoothened. In a randomized phase II study of low dose cytarabine 20 mg given subcutaneously twice daily for 10 days with or without glasdegib 100 mg daily, the glasdegib arm demonstrated a statistically significant improved CR rates (15% versus 2.3%) and OS (8.3 months versus 4.3 months).
Glasdegib was approved by the FDA in combination with low dose cytarabine for unfit AML patients in late 2018 [75]. Two separate studies involving Glasdegib are underway: one with azaciditine for treatment of adult patients with previously untreated AML who are not candidates for intensive induction chemotherapy, and another in combination with standard intensive induction chemotherapy. (clinicaltrials.gov identifier: NCT 03416179)
Bispecific antibodies
Bispecific antibodies have been successfully used in B cell acute lymphoblastic leukemia. They have two different variable regions: one that binds to the T-cell subunit CD3 and other to the tumor surface antigen to provide contact between the cells, creating a T-cell response against tumor cells. Concerns associated with use of bi-specific antibodies in AML include limited efficacy in high tumor burden states, questionable efficacy in patients with extramedullary disease and risk of life-threatening cytokine release syndrome (CRS). AMG330 is a bispecific antibody against CD3 and CD33 that demonstrates AML cell cytotoxicity and has synergy with blockade of the PD-1/PD-L1 axis, thus reversing a T-cell-induced immune escape mechanism [76]. Currently, phase I clinical trials are underway (clinicaltrials.gov identifier: NCT 02520427).
AMV564 is a novel bivalent, bispecific CD33/CD3 T-cell engager that binds CD33 on target AML cells and CD3 on T-cells leading to T-cell-directed lysis of CD33+ leukemic blasts. AMV564 has reduced clearance and has a longer half-life than monovalent, bispecific T-cell engagers. It is currently in phase 1 clinical trial in relapsed /refractory AML. (ClinicalTrials.gov Identifier: NCT03144245). Flotetuzumab is a dual-affinity retargeting antibody (DART) comprising of 2 independent polypeptides fusing the heavy-chain variable domain of one antibody to the light-chain variable domain of the other to connect CD3 and CD123. CD123, the interleukin-3 receptor alpha chain, has been reported to be over-expressed on malignant AML cells. The primary mechanism of action of flotetuzumab is possibly its ability to redirect T lymphocytes to kill CD123-expressing cells. At least 2 phase 1 studies in relapsed/refractory AML are currently in progress. (clinicaltrials.gov identifiers NCT04158739, NCT02152956). An early phase study with flotetuzumab in relapsed/refractory AML demonstrated encouraging responses, including patients with altered TP53 status [77].
A potential area of concern in targeting CD33 has been elucidated. Alternative splicing of CD33 RNA results in shorter isoform expressed on the cell surface, which lacks the V-set but retains the C2-set Ig-like domain. A single nucleotide polymorphism (SNP), rs12459419 (C > T; Ala14Val), was present in ∼50% of North and South American and European AML population and leads to skipping of exon 2 of CD33, which results in deletion of the V domain of CD33 [78]. Several CD33-antibody–based therapies, including gemtuzumab ozogamicin, bind and recognize the V domain of CD33. JNJ-67571244, a human bispecific antibody is capable of binding to the C2 domain of CD33 and to CD3, to induce T-cell recruitment and tumor cell cytotoxicity. It is currently in phase 1 clinical trials to treat patients with relapsed/refractory AML and high-risk MDS (#NCT039153).
Check Point Inhibition
Ipilimumab: Ipilimumab, a CTLA 4 antibody, has been studied in AML patients relapsing post allogenic stem cell transplant. 5 of 12 AML patients in the study had responses, including four patients with extramedullary disease [79]. Ipilimumab is being evaluated with decitabine in a phase 1 study in patients with relapsed/refractory MDS and AML. (Cliniclatrials.gov identifier: NCT 02890329)
CAR-T therapy: CAR-T-cell therapy has been successfully used in B cell non-Hodgkin lymphoma and B cell acute lymphoblastic leukemia. However, it’s progress in AML has been limited due to lack of a consistently expressed target. Lewis-Y antigen, CD 33, CD 123, C-type lectin-like molecule 1 (CLL-1) targeting CAR-T constructs have been in clinical trials. A phase I study of autologous CAR anti-LeY T-cell therapy of AML evaluated safety and post infusion persistence of adoptively transferred T cells in four patients. Two patients achieved stable disease (duration 23 months in 1 patient), and an additional 2 patients had transient response (blasts reduction/cytogenetic remission). No severe adverse events (AEs) or cytokine release syndrome was observed [80].
Other targets: Numerous other targets have been identified, and active trials are underway. These include cell cycle inhibitors like aurora kinase inhibitors, polo like kinase-1 inhibitors, cyclin dependent kinase inhibitors; targeting epigenetic dysregulation, including bromodomain and extra-terminal protein family inhibitors (BET), MLL fusion protein /gene re-arrangement targeting agents; peptide and dendritic cell vaccines.
- Rational combination of therapies
- Sequencing of therapies
- Role and timing of allogenic stem cell transplantation
- Role and duration of maintenance
- Integration of MRD status with timing, duration and type of treatment
Case 1
A very fit 72-year-old male presented to his dentist with gingival hyperplasia and perioral abnormalities concerning for infection leading to referral to the emergency department. He was found to have white blood cell count over 360,000 and bone marrow biopsy was consistent with AML. Cytogenetics were 46,XY. Molecular testing, including next generation sequencing, identified NPM1, CEBPA, TET2, and FLT3-ITD mutations with allelic ratio of 0.4. He received induction chemotherapy plus midostaurin. His induction course was complicated by cardiac arrest due to septic shock, though he fully recovered. He subsequently received consolidation chemotherapy with midostaurin and completed 1 year of midostaurin maintenance. Six weeks after discontinuing midostaurin maintenance, he presented to the emergency department with fatigue and was found to have a white blood cell count of 260,000. Bone marrow biopsy was consistent with relapsed AML with the same molecular profile (though FLT3-ITD allelic ratio had increased to 0.6). He was started on single agent gilteritinib. After four total cycles of gilteritinib (the time of this manuscript), he has achieved morphologic complete remission.
Case 2
A 50-year-old man with history of deep vein thrombosis and obesity was diagnosed with AML during evaluation of pancytopenia. Cytogenetics were 46,XY. Molecular analysis including next generation sequencing for 36 gene mutations identified NPM1 and IDH1 mutations. He received induction chemotherapy and day 14 bone marrow biopsy showed evidence of refractory AML, so he received a second induction chemotherapy after which he attained morphologic remission. The NPM1 mutation was no longer detectable, but IDH1 mutation persisted. Allogeneic stem cell transplant was recommended, but while awaiting an eligible donor, he relapsed. A third attempt at reinduction chemotherapy failed to achieve complete remission, and he began ivosidenib therapy. After one cycle of ivosidenib, he regained morphologic complete remission. He subsequently received an allogeneic bone marrow transplant from an unrelated donor. As of the time of this manuscript, he is one-year post-transplant and remains in complete remission with full donor chimerism, without evidence of either NPM1 or IDH1 mutations.
The treatment paradigm of AML has changed for the first time since the 1970’s (Table 2). Patients now have treatment options in addition to or instead of chemotherapy, improving their overall survival. Targeted treatment is changing prognostic models, since mutations of poor prognosis are subdued with therapy. As we further understand the biology of cell surface markers, cytogenetic and molecular markers that drive AML and promote leukemogenesis, additional targeted therapies are expected. Research is ongoing to develop new agents, new combinations of existing treatments, and develop mechanisms to overcome treatment resistance.
Table 2. Targeted therapy in AML
Target |
Biology |
Treatment |
Indication |
Administration |
FLT3 |
Tyrosine kinase receptor
ITD and TKD mutations lead to ligand-independent phosphorylation and activation |
Midostaurin |
Combination with induction chemotherapy, newly diagnosed AML |
Oral |
Gilteritinib |
Single agent for relapsed/refractory AML |
Oral |
IDH1 |
Mutations impede conversion of isocitrate to α-KG in Krebs cycle; instead generate 2-HG, oncometabolite, that halts cell differentiation and promotes leukemic tumor genesis. |
Ivosidenib |
Relapsed/Refractory AML with IDH1 mutation |
Oral |
IDH2 |
Mutations impede conversion of isocitrate to α-KG in Krebs cycle; instead generate 2-HG, oncometabolite, that halts cell differentiation and promotes leukemic tumor genesis. |
Enasidenib |
Relapsed/Refractory AML with IDH2 mutation |
Oral |
CD33 |
Transmembrane receptor expressed by AML blasts |
Gemtuzumab ozogamicin |
Combination with induction chemotherapy, newly diagnosed AML
Single agent, newly diagnosed AML
Single agent, relapsed/refractory AML |
Intravenous |
- Ley TJ (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059-2074.
- Döhner H (2017) Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129: 424-447. [Crossref]
- Tallman MS (2019) Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 17: 721-749. [Crossref]
- National Cancer Institute Surveillance, Epidemiology, and End Results. [cited 2020 June 14]; Available from: https://seer.cancer.gov/statfacts/html/amyl.html.
- Santos FP (2011) Prognostic value of FLT3 mutations among different cytogenetic subgroups in acute myeloid leukemia. Cancer 117: 2145-2155. [Crossref]
- Kottaridis PD, Gale RE, Linch DC (2003) Flt3 mutations and leukaemia. Br J Haematol 122: 523-538. [Crossref]
- Lagunas-Rangel FA, Chávez-Valencia V (2017) FLT3-ITD and its current role in acute myeloid leukaemia. Med Oncol 34: 100-114. [Crossref]
- Stirewalt DL, Radich JP (2003) The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer 3: 650-665. [Crossref]
- Levis MD (2003) Small, FLT3: ITDoes matter in leukemia. Leukemia 17: 1738-1752. [Crossref]
- Bacher U (2008) Prognostic relevance of FLT3-TKD mutations in AML: the combination matters--an analysis of 3082 patients. Blood 111: 2527-2537. [Crossref]
- Mead AJ (2007) FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood 110: 1262-1270. [Crossref]
- Fischer T (2010) Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol 28: 4339-4345. [Crossref]
- Stone RM (2012) Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia 26: 2061-2068. [Crossref]
- Stone RM (2017) Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 377: 454-464.
- Jaramillo S (2017) Condensed versus standard schedule of high-dose cytarabine consolidation therapy with pegfilgrastim growth factor support in acute myeloid leukemia. Blood Cancer J 7: e564. [Crossref]
- Perl AE (2017) Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol 18: 1061-1075. [Crossref]
- Perl AE (2019) Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med 381: 1728-1740. [Crossref]
- Ohanian M (2018) Sorafenib Combined with 5-azacytidine in Older Patients with Untreated FLT3-ITD Mutated Acute Myeloid Leukemia. Am J Hematol 93: 1136-1141. [Crossref]
- Daver N (2019) Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia 33: 299-312. [Crossref]
- Randomized Trial of Gilteritinib vs Midostaurin in FLT3 Mutated Acute Myeloid Leukemia. [cited 2020 June 13]; Available from: ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03836209.
- A Study of CPX-351 (Vyxeos) with Quizartinib for the Treatment of FLT3-ITD Mutation-Positive Acute Myeloid Leukemia. [cited 2020 June 13]; Available from: ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04209725.
- Cortes JE (2019) Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol 20: 984-997. [Crossref]
- Study of crenolanib vs midostaurin following induction chemotherapy and consolidation therapy in newly diagnosed flt3 mutated aml. [cited 2020 June 13]; Available from: https://clinicaltrials.gov/ct2/show/NCT03258931.
- A Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients with FLT3 Activating Mutations [cited 2020 June 13]; Available from: ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01657682.
- Reitman ZJ, Yan H (2010) Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst 102: 932-941. [Crossref]
- Rakheja D (2013) The emerging role of d-2-hydroxyglutarate as an oncometabolite in hematolymphoid and central nervous system neoplasms. Front Oncol 3: 169. [Crossref]
- Dang L (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462: 739-744. [Crossref]
- DiNardo CD (2016) A phase I Study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations. Blood 128: 1073-1073.
- Im AP (2014) DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia 28: 1774-1783. [Crossref]
- Green CL (2011) The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood 118: 409-412. [Crossref]
- Patel JP (2012) Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 366: 1079-1089. [Crossref]
- Papaemmanuil E (2016) Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med 374: 2209-2221. [Crossref]
- Paschka P (2010) IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 28: 3636-3643. [Crossref]
- Kim ES (2017) Enasidenib: First Global Approval. Drugs 77: 1705-1711. [Crossref]
- Dhillon S (2018) Ivosidenib: First Global Approval. Drugs 78: 1509-1516. [Crossref]
- Roboz GJ (2020) Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 135: 463-471. [Crossref]
- Norsworthy KJ (2020) Differentiation syndrome with ivosidenib and enasidenib treatment in patients with relapsed or refractory IDH-mutated AML: a U.S. Food and Drug Administration systematic analysis. Clinical Cancer Research 0834.2020.
- Amatangelo MD (2017) Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 130: 732-741. [Crossref]
- Stein EM (2017) Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130: 722-731. [Crossref]
- Buege MJ, DiPippo AJ, DiNardo CD (2018) Evolving treatment strategies for elderly leukemia patients with IDH mutations. Cancers (Basel)10. [Crossref]
- Konopleva M (2016) Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov 6: 1106-1117. [Crossref]
- Kung Sutherland MS (2013) SGN-CD33A: a novel CD33-targeting antibody–drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 122: 1455-1463. [Crossref]
- Zhao L (2019) CD33 in alzheimer's disease - biology, pathogenesis, and therapeutics: A mini-review. Gerontology 65: 323-331. [Crossref]
- Bross PF (2001) Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res 7: 1490-1496. [Crossref]
- Petersdorf SH (2013) A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 121: 4854-4860. [Crossref]
- Institute NC (2010) Gemtuzumab ozogami- cin voluntarily withdrawn from U.S. Markets: National Cancer Institute. Retrieved from https://www.cancer. gov/about-cancer/treatment/drugs/fda-gemtuzumab- ozogamicin
- Administration FDA news release (2017) FDA approves Mylotarg for treatment of acute myeloid leukemia. Retrieved from https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm574507.htm. 2017.
- Castaigne S (2012) Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 379: 1508-1516. [Crossref]
- Selby C, Yacko LR, Glode AE (2019) Gemtuzumab ozogamicin: Back again. J Adv Pract Oncol 10: 68-82. [Crossref]
- Appelbaum FR, Bernstein ID (2017) Gemtuzumab ozogamicin for acute myeloid leukemia. Blood 130: 2373-2376. [Crossref]
- Lo-Coco F (2004) Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood 104: 1995-1999. [Crossref]
- Hart S (2011) SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia 25: 1751-1759. [Crossref]
- Konopleva M, Letai A (2018) BCL-2 inhibition in AML: an unexpected bonus? Blood 132: 1007-1012. [Crossref]
- DiNardo CD (2018) Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol 19: 216-228. [Crossref]
- DiNardo CD (2019) Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 133: 7-17. [Crossref]
- Wei A (2018) Venetoclax with low-dose cytarabine induces rapid, deep, and durable responses in previously untreated older adults with aml ineligible for intensive chemotherapy. Blood. 132: 284-284.
- A HW (2020) A phase 3 study of venetoclax plus low dose cytarabine in previously untreated older patients with Acute Myeloid Leukemia (VIALE- C): A 6 month update.EHA.
- EHA (2020) A study of venetoclax in combination with azacitidine versus azacitidine in treatment naïve subjects with acute myeloid leukemia who are ineligible for standard induction therapy.
- Curtis Andrew Lachowiez GB, Sanam L, Zhihong Z, Kadia TM, Masarova L, et al. (2020) Phase Ib/II study of the IDH1-mutant inhibitor ivosidenib with the BCL2 inhibitor venetoclax +/- azacitidine in IDH1-mutated hematologic malignancies. J Clin Oncol 38.
- Luedtke DA (2017) Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct Target Ther 2: 17012. [Crossref]
- Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307-310. [Crossref]
- Barbosa K (2019) The role of TP53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosomes Cancer 58: 875-888. [Crossref]
- Kadia TM (2016) TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 122: 3484-3491. [Crossref]
- Sallman DA (2019) Phase 2 results of APR-246 and azacitidine (AZA) in patients with TP53 mutant myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia (AML). Blood 134: 676-676.
- Cluzeau T (2019) APR-246 combined with azacitidine (AZA) in TP53 mutated myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). a Phase 2 study by the group efrancophone desyélodysplasies (GFM). Blood 134: 677-677.
- APR-246 in Combination with Venetoclax and Azacitidine in TP-53 Mutated Myeloid Malignancies. [cited 2020 June 13]; Available from: https://clinicaltrials.gov/ct2/show/NCT04214860.
- APR-246 in Combination with Azacitidine for TP-53 Mutated AML (Acute Myeloid Leukemia) or MDS (Myelodysplastic Syndrome) Following Allogeneic Stem Cell Transplant. [cited 2020 June 13]; Available from: https://clinicaltrials.gov/ct2/show/NCT03931291.
- APR-246 Combined with Azacitidine in TP53 Mutated Myelodysplastic Syndromes (MDS) and Acute Myeloid Leukemia. A Phase 2 Study by the Groupe Francophone des Myélodysplasies (GFM). 2020: EHA.
- Erba HP (2019) Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv 3: 1939-1949. [Crossref]
- Rashidi A, Uy GL (2015) Targeting the microenvironment in acute myeloid leukemia. Curr Hematol Malig Rep 10: 126-131. [Crossref]
- DeAngelo DJ (2016) A phase I/II study of GMI-1271, a novel e-selectin antagonist, in combination with induction chemotherapy in relapsed/refractory and elderly previously untreated acute myeloid leukemia; results to date. Blood 128: 4049-4049.
- DeAngelo DJ (2017) GMI-1271, a novel E-selectin antagonist, combined with induction chemotherapy in elderly patients with untreated AML. Journal of Clinical Oncology 35: 2560-2560.
- Ingham PW (2011) Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet pp: 303-406. [Crossref]
- Queiroz KC (2010) Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 29: 6314-6322. [Crossref]
- Cortes JE (2016) A phase 2 randomized study of low dose Ara-C with or without Glasdegib (PF-04449913) in untreated patients with acute myeloid leukemia or high-risk myelodysplastic syndrome. Blood 128: 99-99.
- Harrington KH (2015) The broad anti-AML activity of the CD33/CD3 BiTE antibody construct, amg 330, is impacted by disease stage and risk. PLoS One 10: e0135945. [Crossref]
- Jayakumar Vadakekolathu CL, Stephen Reeder, Sarah E Church, Tressa Hood, John Muth, et al. (2020) TP53 abnormalities correlate with immune infiltration and are associated with response to flotetuzumab, an investigational immunotherapy,in acute myeloid leukemia. 2020: ACR Virtual Annual Meeting 2020 April 27-28, 2020 Clinical Plenary – Abstract #9670.
- Lamba JK (2017) CD33 splicing polymorphism determines gemtuzumab ozogamicin response in de novo acute myeloid leukemia: Report from randomized phase III children's oncology group trial AAML0531. J Clin Oncol 35: 2674-2682. [Crossref]
- Davids MS (2016) Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med 375: 143-153. [Crossref]
- Ritchie DS (2013) Persistence and efficacy of second-generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther 21: 2122-2129. [Crossref]