The isoforms of the APOE gene are of profound importance regarding the onset of Alzheimer’s disease (AD), with APOE2 conferring resistance, APOE3 conferring neutral susceptibility, and APOE4 conferring proneness to AD. L-cysteine is an amino acid that has several anti-AD properties, among which are its ability to sequester iron and form glutathione (GSH), a powerful antioxidant. In our experiment, we fed Mus musculus (mice) homozygous for APOE2, APOE3, and APOE4 either a control diet or a diet high in L-cysteine. Using Western blotting analysis, we quantified total APOE proteins extracted from post-mortem brains of APOE2, APOE3, and APOE4 homozygous mice, total Amyloid β (Aβ) protein, and total hyper-phosphorylated Tau (HP-Tau) from mice at 3-, 6-, 9-, and 12-month ages. We found that administration of L-cysteine trends toward lowering levels of Aβ in the APOE3 cohort, but this effect is statistically insignificant. On the other hand, L-cysteine caused a significant increase in APOE4 abundance, but a significant decrease in APOE3 abundance regarding diet [F(6,42) = 5.61, p = 0.01]. Furthermore, administration of L-cysteine revealed trends toward lowering HP-Tau deposition in the APOE2 and APOE3 cohorts, but increasing deposition in the APOE4 cohort, although these effects are statistically insignificant. Moreover, immunohistochemistry analyses on the hippocampus and midsagittal brain revealed no effects of L-cysteine on Aβ. Results also showed a decrease in HP-Tau without regard to APOE genotype, but this was not statistically significant (p = 0.18). Taken together, these data suggest that L-cysteine may serve as a promising intervention for AD pathology, although future studies necessitate increasing statistical power to confirm the effect of diet on Aβ and HP-Tau deposition.
APOE, L-Cysteine, Amyloid β, Tau
L-cysteine shows promise as a dietary method of helping to treat APOE4-induced AD. Currently, it is known that a triple-combination diet of N-acetylcysteine (NAC – a prodrug to L-cysteine), acetyl-L-carnitine (ALCAR), and S-adenosylmethionine (SAMe) administered to APOE4 mice almost completely reverses AD symptoms [1]. However, in humans, NAC and SAMe have mild and potentially severe adverse effects. For a safer method, we used L-cysteine in place of NAC in this experiment to assess the individual contribution of NAC to the triple-combination diet. Taken together, this experiment reveals further knowledge and understanding about utilizing L-cysteine as a potentially safe and efficient way of combating APOE4-induced AD pathology.
Several factors are involved in the onset and regulation of AD. Among these factors are the APOE gene family and its protein products. One function of the APOE protein is clearing Aβ from the brain, where it works by co-localizing with Aβ after it is secreted into the perivascular space [2], thereby resulting in a decrease in oxidative stress and AD symptoms [3]. A major role of Aβ is to sequester iron, which is toxic to the brain [4]. Moreover, Aβ can accumulate in the extracellular space and form plaques, which are toxic to neurons and thereby, facilitate the onset of AD.
There are three different alleles for APOE: ε2, ε3, and ε4 – each of which codes for a protein that differs at only one or two amino acids from and in the same positions as the others (Tables 1 and 2) [5]. Thus, there are six different genotypes of APOE that exist naturally: APOE2, APOE2/3, APOE2/4, APOE3, APOE3/4, and APOE4. Those with the APOE2 genotype are at the most reduced risk for AD, because the ε2 allele has protective effects against AD [6]. Consequently, AD risk is reduced somewhat for APOE2/3, neutral for APOE2/4 and APOE3, elevated for APOE3/4, and greatest for APOE4 [7].
Table 1. Amino Acid Differences among APOE Isoforms
APOE Isoform |
Amino Acid Differences |
Number of Tyrosines |
APOE2 |
Cys-112; Cys-158 |
4 |
APOE3 |
Cys-112; Arg-158 |
4 |
APOE4 |
Arg-112; Arg-158 |
4 |
Table 2. Amino Acid Sequences of APOE2, APOE3, APOE4, and HtrA1. Arginines are signified by the letter “R”. Cysteines are signified by the letter “C”. Tyrosines are signified by the letter “Y”
Protein Isoform |
Protein Sequence |
APOE2 |
MKVEQAVETE PEPELRQQTE WQSGQRWELA LGRFWDYLRW VQTLSEQVQE ELLSSQVTQE LRALMDETMK ELKAYKSELE EQLTPVAEET RARLSKELQA AQARLGADME DVCGRLVQYR GEVQAMLGQS TEELRVRLAS HLRKLRKRLL RDADDLQKCL AVYQAGAREG AERGLSAIRE RLGPLVEQGR VRAATVGSLA GQPLQERAQA WGERLRARME EMGSRTRDRL DEVKEQVAEV RAKLEEQAQQ IRLQAEAFQA RLKSWFEPLV EDMQRQWAGL VEKVQAAVGT SAAPVPSDNH |
APOE3 |
MKVEQAVETE PEPELRQQTE WQSGQRWELA LGRFWDYLRW VQTLSEQVQE ELLSSQVTQE LRALMDETMK ELKAYKSELE EQLTPVAEET RARLSKELQA AQARLGADME DVCGRLVQYR GEVQAMLGQS TEELRVRLAS HLRKLRKRLL RDADDLQKRL AVYQAGAREG AERGLSAIRE RLGPLVEQGR VRAATVGSLA GQPLQERAQA WGERLRARME EMGSRTRDRL DEVKEQVAEV RAKLEEQAQQ IRLQAEAFQA RLKSWFEPLV EDMQRQWAGL VEKVQAAVGT SAAPVPSDNH |
APOE4 |
MKVEQAVETE PEPELRQQTE WQSGQRWELA LGRFWDYLRW VQTLSEQVQE ELLSSQVTQE LRALMDETMK ELKAYKSELE EQLTPVAEET RARLSKELQA AQARLGADME DVRGRLVQYR GEVQAMLGQS TEELRVRLAS HLRKLRKRLL RDADDLQKRL AVYQAGAREG AERGLSAIRE RLGPLVEQGR VRAATVGSLA GQPLQERAQA WGERLRARME EMGSRTRDRL DEVKEQVAEV RAKLEEQAQQ IRLQAEAFQA RLKSWFEPLV EDMQRQWAGL VEKVQAAVGT SAAPVPSDNH |
HtrA1 |
MQIPRAALLP LLLLLLAAPA SAQLSRAGRS APLAAGCPDR CEPARCPPQP EHCEGGRARD ACGCCEVCGA PEGAACGLQE GPCGEGLQCV VPFGVPASAT VRRRAQAGLC VCASSEPVCG SDANTYANLC QLRAASRRSE RLHRPPVIVL QRGACGQGQE DPNSLRHKYN FIADVVEKIA PAVVHIELFR KLPFSKREVP VASGSGFIVS EDGLIVTNAH VVTNKHRVKV ELKNGATYEA KIKDVDEKAD IALIKIDHQG KLPVLLLGRS SELRPGEFVV AIGSPFSLQN TVTTGIVSTT QRGGKELGLR NSDMDYIQTD AIINYGNSGG PLVNLDGEVI GINTLKVTAG ISFAIPSDKI KKFLTESHDR QAKGKAITKK KYIGIRMMSL TSSKAKELKD RHRDFPDVIS GAYIIEVIPD TPAEAGGLKE NDVIISINGQ SVVSANDVSD VIKRESTLNM VVRRGNEDIM ITVIPEEIDP |
In addition, a relationship exists between the clearance of Aβ and the isoformic identity of APOE. Aβ clearance decreases from APOE2 to APOE3 to APOE4 [8]. Moreover, due to the presence of the cysteine residue in APOE3, disulfide bridges can form between APOE3 molecules, allowing APOE3 to exist as monomers and homodimers [9]. Since APOE2 has two cysteine residues, it can exist not only as monomers and homodimers, but also as homopolymers [9]. Conversely, the lack of cysteine residues in APOE4 does not allow for the formation of disulfide bonds between APOE4 molecules, which may contribute to its inefficiency at clearing Aβ. The resulting buildup of Aβ increases oxidative stress and further facilitates the onset of AD in APOE4 genotypes [10]. Another problem with APOE4 is that it inhibits the degradation of HP-Tau in vitro [11]. However, APOE4 is degraded to a greater extent in the brain by the HtrA1 protein than is APOE3 in vitro [11], thus resulting in somewhat of a paradox between its effects on Aβ and HP-Tau clearance.
Another factor that facilitates the onset of AD is the protein Tau, which is necessary for the integrity of microtubules. Under degenerative conditions, mechanisms such as mitochondrial and metabolic dysfunction, as well as loss of metal homeostasis, result in the formation of reactive oxygen species (ROS) that cause hyper-phosphorylation of Tau [12]. Normally, HP-Tau proteins are degraded and cleared by proteosomes, but in AD patients, the high levels of Aβ inhibit these proteosomes, leading to the over-proliferation of HP-Tau [13]. This over-proliferation contributes to the formation of neurofibrillary tangles, which in turn increases oxidative stress [12]. Thus, APOE4, Aβ plaques, and HP-Tau tangles facilitate the onset of AD pathology.
Aside from the genetics of AD, there has also been research on how diet influences AD symptoms caused by APOE4. Specifically, researchers Chan, et al. (2008) tested a triple-combination diet consisting of NAC, ALCAR, and SAMe on the behavioral function of APOE4 mice [1]. The results showed a significant increase in behavioral function. For example, attack latency improved by approximately 90%, attack frequency decreased about 95%, and Y-maze testing showed that cognitive function increased by about 30% and then after one month, up to 70% [1]. Therefore, the extents of the individual roles of L-cysteine, ALCAR, and SAMe in treating AD in APOE4 patients should be researched.
Sources of reagents, antibodies, and proteins
Protease inhibitor and rabbit polyclonal anti-phospho-MAPT (pSer262) primary antibodies were purchased from Sigma-Aldrich. BCA assay kit was purchased from ThermoFisher Scientific (Pierce) and Spectra multicolor broad range protein ladder was purchased from ThermoFisher Scientific. Precast Criterion Tris-HCl protein gels and nitrocellulose membranes were purchased from Bio-Rad. Rabbit polyclonal anti-APOE2/APOE3/APOE4 primary antibodies were purchased from BioVision. Rabbit polyclonal anti-beta Amyloid 1-42 primary antibodies were purchased from Bioss Antibodies. Rabbit monoclonal anti-Vinculin primary antibodies were purchased from Cell Signaling Technology. IRDye 800CW donkey anti-rabbit secondary antibodies were purchased from Li-Cor. Purified (>97%) mouse Amyloid 1-42 protein was purchased from rPeptide: Premiere Peptide Solutions. Purified (>95%) human MAPT protein was purchased from Syd Labs. Purified (>95%) human APOE3 and APOE4 (>90%) proteins were purchased from ProSpec Protein Specialists. Purified (>90%) human APOE2 was purchased from Creative BioMart.
Source of transgenic mice
APOE4-tg knockout mice (Tg(GFAP-APOE*4)1Hol) for endogenous mouse APOE but expressing human APOE4 under the direction of the human glial fibrillary acidic protein (GFAP) promoter, were purchased from Jackson Laboratory, USA. APOE3-tg knockout mice (Tg(GFAP-APOE*3)37Hol) for endogenous mouse APOE but expressing human APOE3 under the direction of the human GFAP promoter, were purchased from Jackson Laboratory, USA. APOE2-tg knockout mice (Tg(GFAP-APOE*2)14Hol) for endogenous mouse APOE but expressing human APOE2 under the direction of the human GFAP promoter, were purchased from Jackson Laboratory, USA. APOE genotypes were not mixed when breeding these mice or any progeny mice in order to keep all mice homozygous with respect to APOE genotype. APOE3 homozygotes were considered wild type. Breeders were kept separate from experimental mice, and both male and female mice were used in this experiment.
Sources of control and experimental diets
The control diet was purchased from Envigo and consisted of standard laboratory chow, which contained 4g L-cysteine/kg total food weight. L-cysteine (97% purity) was purchased from Sigma-Aldrich. To make the experimental diet, 10g L-cysteine/kg total food weight was added to the control diet by Envigo, along with a blue-green dye for ease in identification. Both diets were isocaloric in relation to each other and were of equal palatability.
Care of mice
Experimental mice were divided into control and experimental diet groups. Prior to 2 months of age, the experimental diet group was fed the control diet. From 2 months of age until being sacrificed, the experimental diet group was fed only the experimental diet. The control diet group was always fed the control diet. All animals were housed under standard conditions (20 ̊C temperature, 12-hour light-dark cycle) with equal free access to air, food, and water. All procedures were performed in compliance with the USDA Animal Welfare Act and the NIH Public Health Service Policy on the Humane Care and Use of Animals.
Tissue preparation
In compliance with institutional guidelines for the humane treatment of animals, mice were sacrificed using chambers containing isofluorane gas at 3-months, 6-months, 9-months, and 12-months of age. Mouse brains were carefully removed and cut in the mid-sagittal plane. For each brain, one hemisphere was snap frozen at -80 ̊C for Western blot analysis and the other hemisphere was fixed in 4% formaldehyde solution for immunohistochemistry analysis. Each frozen sample was thawed and homogenized in a mixture of protease inhibitor (1:100 dilution) and 25mM ammonium bicarbonate; following which, BCA analyses were performed.
Levels of APOE, Aβ, and HP-Tau proteins were examined by Western blotting in all 48 mice. For each sample, ~30μg homogenate protein was separated using precast 4-15% and 4-20% gradient SDS-PAGE gels and transferred to nitrocellulose membranes (pore sizes 0.45μm). Each gel also contained ~2μg of a purified control protein based off of which primary antibody was to be used on its respective blot. All gel loading volumes were equal. Each membrane was incubated with its respective primary antibody (anti-APOE2/APOE3/APOE4, 1:3000; anti-beta Amyloid 1-42, 1:2000; anti-phospho-MAPT (pSer262), 1:1000; anti-Vinculin, 1:1000) in blocking buffer (3% nonfat dried milk in 1× tris-buffered saline with Tween 20 (TBST)) overnight at 4 ̊C, washed three times with 1× TBST, and then incubated in blocking buffer for 1 hour at room temperature with Li-Cor IRDye 800CW secondary antibodies (donkey anti-rabbit IgG, 1:10000 dilution). Levels of proteins were estimated by densitometry analysis using the Li-Cor Image Studio 4.0 software. Anti-Vinculin immunoblots were used as loading controls.
Western blot statistical analysis
Western blot images were quantified using the Li-Cor Image Studio 4.0 software and each image was cleaned three times using the noise removal tool. Aβ bands were selected for quantification at ~45kD, APOE bands at ~34kD, and HP-Tau bands at ~62kD. Selected bands were quantified based on their relative intensities and adjusted for background. Statistical tests for the comparison of diet palatability were performed using SPSS v25 (IBM, Inc.), where differences in mouse weight between diet type and genotype cohorts were analyzed using a 2x2 factorial ANOVA statistical model. We also performed a factorial ANOVA for relationships between diet type, APOE genotype, sex, and age, each with the amount of APOE, Aβ, and HP-Tau in the gels, with a Bonferroni correction for a posteriori comparisons of pairwise comparisons. Statistical significance was defined at α < 0.05. Trending toward statistical significance was defined by non-overlapping standard error bars with 0.05 < α < 0.10.
For a subset of mice equally distributed across cohorts, with brain hemispheres preserved in formalin, we stained for the presence of Aβ (21 mice) and HP-Tau (39 mice). Due to technical difficulties with staining, we were not able to perform immunohistochemistry for the presence of APOE.
Brain hemispheres were paraffin-processed and sliced in the sagittal orientation on a rotary microtome at 6 microns. The first two serial mid-sagittal sections exhibiting the extent of rostral-to-caudal anatomy were chosen for immunohistochemistry staining.
Tissue samples were incubated for one hour at 37°C before undergoing deparaffinization and rehydration. A solution of 3% hydrogen peroxide/peroxidase was applied to each tissue to remove any endogenous enzymatic activity. Antigen retrieval was completed by immersing the samples in Diva Decloaker 10X (1:10, BioCare Medical, Pacheco, CA) at 95°C for 20 minutes. Nonspecific antibody site blocking was completed using two 10-minute washes with tris-buffered saline/Polysorbate 20 (TBST) followed by incubation in goat serum at room temperature for 15 minutes. Each serial sample was then covered with an unconjugated polyclonal antibody to either anti-phospho-MAPT (pSer262) (1:200, Sigma-Aldrich, St. Louis, MO) or anti-beta Amyloid 1-42 (1:500, Sigma-Aldrich, St. Louis, MO), and incubated for one hour at room temperature. Following primary antibody staining, sections were treated with MACH 2 Universal HRP-Polymer Detection (BioCare Medical, Pacheco, CA) and incubated for 30 minutes at room temperature. Diaminobenzidine (DAB) was used as the chromogen and each section was then counterstained with hematoxylin. Following counterstaining, each slide was dehydrated and mounted.
All slides were digitally imaged using a Leica Biosystems Aperio AT2 scanner (20Xz objective lens, Leica Biosystems, Inc., Buffalo Grove, IL) by imaging technologists at LUMEA (Lehi, UT). Digital slides were then uploaded into Aperio ImageScope (v12.3.2.8013, Leica Biosystems, Inc., Buffalo Grove, IL). Using ImageScope, the entire brain slice and hippocampus were selected as regions of interest (ROI) and the area was calculated for each. Stain intensity within each ROI was analyzed via the Positive Pixel Count v9 (PPCv9) algorithm. Input settings for the analysis were modified to apply tighter guidelines for the classification of potential background staining. The sum of positive and strong positive pixels was normalized to each ROI area, and these normalized values were used for statistical analysis.
Immunohistochemistry statistical analysis
We performed a factorial ANOVA analysis using SPSS v25 (IBM, Inc.) with diet, APOE genotype, sex, and age as factors for the positive pixel count of Aβ and HP-Tau within the entire midsagittal brain section and hippocampus, as separate dependent variables. A post hoc Bonferroni correction was performed to determine pairwise comparisons. Statistical significance was defined at α < 0.05. Trending toward statistical significance was defined by non-overlapping standard error bars with 0.05 < α < 0.10.
Comparison of diet palatability
The difference in palatability between the control and L-cysteine diets was insignificant [F(5,106) = 1.92, p = 0.10], as were the main effects of diet [F(1,106) = 0.78, n = 112 mice, p = 0.38], genotype [F(2,106) = 1.52, p = 0.22], and diet X genotype [F(2,106) = 2.73, p = 0.07].
Comparison of sex
ANOVA analysis revealed no significant effects attributed by sex [F(3,21) = 0.646, p = 0.59].
Western Blot
Effect of L-Cysteine on APOE Abundance
Factorial ANOVA analysis revealed significant main effects on APOE abundance for the following: APOE genotype [F(2,23) = 8.33, p = 0.002], age [F(3,23) = 20.60, p < 0.0001], and APOE genotype X diet [F(2,23) = 5.61, p = 0.01].
Follow-up a posteriori analyses for genotype showed significant increases in APOE abundance between APOE2 and APOE4 genotypes (p = 0.005) and APOE3 and APOE4 genotypes (p = 0.006).
Follow-up a posteriori analyses for age showed significant increases between 3-months and 6-months (p = 0.003), 3-months and 12-months (p = 0.005), and 9-months and 12-months (p < 0.0001); and a significant decrease in APOE abundance between 6-months and 9-months (p < 0.0001).
Although the weight of APOE is ~34kD per pure monomer, APOE2 is known to form monomers, dimers, and polymers of itself; whereas APOE3 is known to form monomers and dimers of itself [9,14]. Furthermore, all APOE isoforms undergo intensive modifications of varying degrees, such as lipidation [15], glycosylation [9,16-20], nitrosylation [21], and sialylation [20,22], as well as form complexes with other proteins, such as Aβ [23]. Moreover, APOE is degraded by the HtrA1 protein into 25-kD fragments in vitro [24]. Thus, not only was APOE seen at ~34kD, but also at ~45kD and ~25kD (Figure 1).
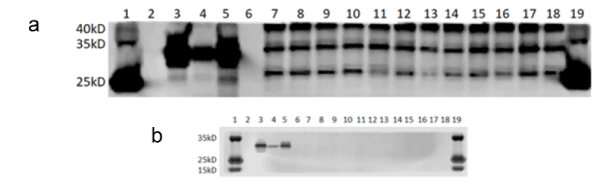
Figure 1. Western Blots Showing APOE Abundance. (a) APOE in complex with Aβ is shown at ~45kD. Non-fragmented APOE and purified APOE protein are shown at ~34kD. APOE fragments are shown at ~25kD. (b) Adjusted lighting reveals clean purified protein and protein molecular weight ladder bands, however eliminating all other bands. (a and b) Lanes are listed in numerical order, from left to right: Lane 1 = protein molecular weight ladder. Lane 2 = empty. Lane 3 = purified human APOE2. Lane 4 = purified human APOE3. Lane 5 = purified human APOE4. Lane 6 = empty. Lane 7 = APOE2/6-month/L-cysteine diet/female. Lane 8 = APOE2/6-month/L-cysteine diet/male. Lane 9 = APOE2/6-month/control diet/female. Lane 10 = APOE2/6-month/control diet/male. Lane 11 = APOE3/6-month/L-cysteine diet/female. Lane 12 = APOE3/6-month/L-cysteine diet/male. Lane 13 = APOE3/6-month/control diet/female. Lane 14 = APOE3/6-month/control diet/male. Lane 15 = APOE4/6-month/L-cysteine diet/female. Lane 16 = APOE4/6-month/L-cysteine diet/male. Lane 17 = APOE4/6-month/control diet/female. Lane 18 = APOE4/6-month/control diet/male. Lane 19 = protein molecular weight ladder
There was a statistically significant increase in APOE4 abundance, a statistically significant decrease in APOE3 abundance, and no effect on APOE2 abundance seen with the L-cysteine diet (Figure 2). Additionally, it is clear from the Western blot image that the L-cysteine diet resulted in visibly less APOE4 fragments than did the control diet (Figure 2).
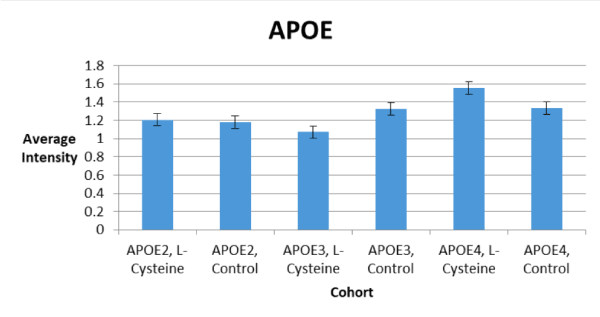
Figure 2. Trends in APOE Abundance
Effect of L-Cysteine on Aβ Abundance
Factorial ANOVA analysis revealed significant main effects on Aβ deposition for the following: age [F(3,23) = 3.77, p = 0.03] and APOE genotype [F(2,23) = 3.35, p = 0.05].
Follow-up a posteriori analyses for age showed a significant increase in Aβ deposition between 9-months and 12-months (p = 0.03).
Follow-up a posteriori analyses for APOE genotype showed a trend toward a significant increase in Aβ deposition between APOE2 and APOE4 (p = 0.06).
Aβ plaques were found at ~45kD (Figure 3). The bands at ~45kD typically represent chains of Aβ bound to a monomer of APOE [23].
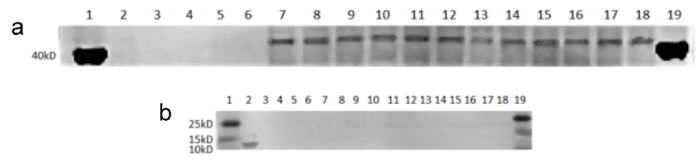
Figure 3. Western Blots Showing Aβ Deposition. (a) Aβ in complex with APOE is shown at ~45kD. (b) Purified Aβ (1-42) protein triplets are shown at ~13kD. (a and b) Lanes are listed in numerical order, from left to right: Lane 1 = protein molecular weight ladder. Lane 2 = purified mouse Aβ (1-42) protein. Lanes 3 – 6 = empty. Lane 7 = APOE2/6-month/L-cysteine diet/female. Lane 8 = APOE2/6-month/L-cysteine diet/male. Lane 9 = APOE2/6-month/control diet/female. Lane 10 = APOE2/6-month/control diet/male. Lane 11 = APOE3/6-month/L-cysteine diet/female. Lane 12 = APOE3/6-month/L-cysteine diet/male. Lane 13 = APOE3/6-month/control diet/female. Lane 14 = APOE3/6-month/control diet/male. Lane 15 = APOE4/6-month/L-cysteine diet/female. Lane 16 = APOE4/6-month/L-cysteine diet/male. Lane 17 = APOE4/6-month/control diet/female. Lane 18 = APOE4/6-month/control diet/male. Lane 19 = protein molecular weight ladder
There were no statistically significant differences in Aβ deposition between the control and L-cysteine diets for any of the cohorts, but there was a trend toward significance for a decrease in Aβ deposition seen in the APOE3 cohort on the L-cysteine diet (Figure 4). It is possible that increasing the number of subjects in the study would provide a better indication of the effect of diet.
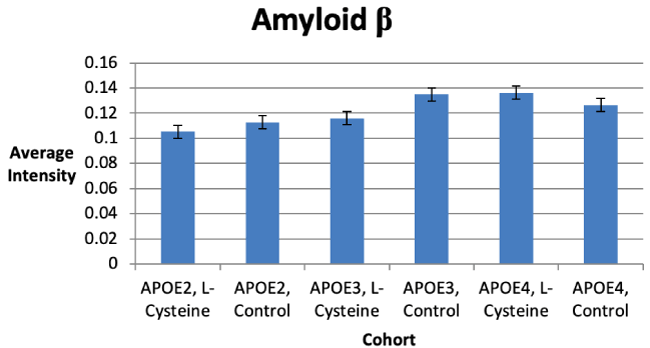
Figure 4. Trends in Aβ Deposition
Effect of L-Cysteine on HP-Tau Abundance
Factorial ANOVA analysis revealed a significant main effect on HP-Tau deposition for age [F(3,23) = 22.90, p < 0.0001].
Follow-up a posteriori analyses for age showed a significant increase between 3-months and 6-months (p < 0.0001); and significant decreases in HP-Tau deposition between 6-months and 9-months (p < 0.0001) and 6-months and 12-months (p < 0.0001).
We consistently saw HP-Tau tangles at 62kD, as was expected (Figure 5). However, there were no statistically significant differences in HP-Tau deposition between the control and L-cysteine diets for any of the cohorts. Nevertheless, we did observe trends toward significance for decreases in HP-Tau deposition seen in the APOE2 and APOE3 cohorts, but for an increase in HP-Tau deposition seen in the APOE4 cohort (Figure 6). It is possible that increasing the number of subjects in the study would provide a better indication of the effect of diet.

Figure 5. Western Blots Showing HP-Tau Deposition. HP-Tau is shown at ~62kD. Purified MAPT is shown at ~46kD and ~64kD. Lanes are listed in numerical order, from left to right. Lane 1 = protein molecular weight ladder. Lane 2 = purified human MAPT protein. Lane 3 = empty. Lane 4 = APOE2/6-month/L-cysteine diet/female. Lane 5 = APOE2/6-month/L-cysteine diet/male. Lane 6 = APOE2/6-month/control diet/female. Lane 7 = APOE2/6-month/control diet/male. Lane 8 = APOE3/6-month/L-cysteine diet/female. Lane 9 = APOE3/6-month/L-cysteine diet/male. Lane 10 = APOE3/6-month/control diet/female. Lane 11 = APOE3/6-month/control diet/male. Lane 12 = APOE4/6-month/L-cysteine diet/female. Lane 13 = APOE4/6-month/L-cysteine diet/male. Lane 14 = APOE4/6-month/control diet/female. Lane 15 = APOE4/6-month/control diet/male. Lane 16 = protein molecular weight ladder
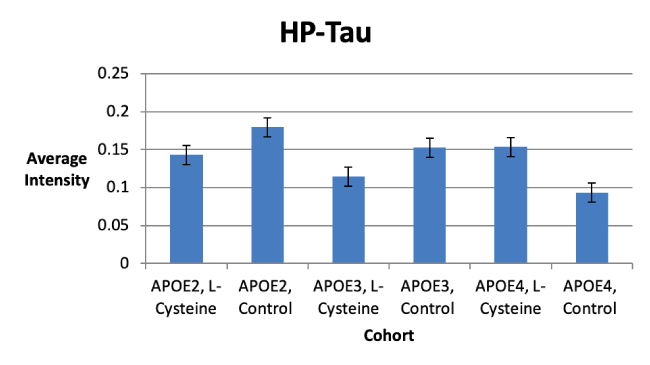
Figure 6. Trends in HP-Tau Deposition
Example images of Aβ and HP-Tau positive pixel count segmentations are shown in Figure 7.
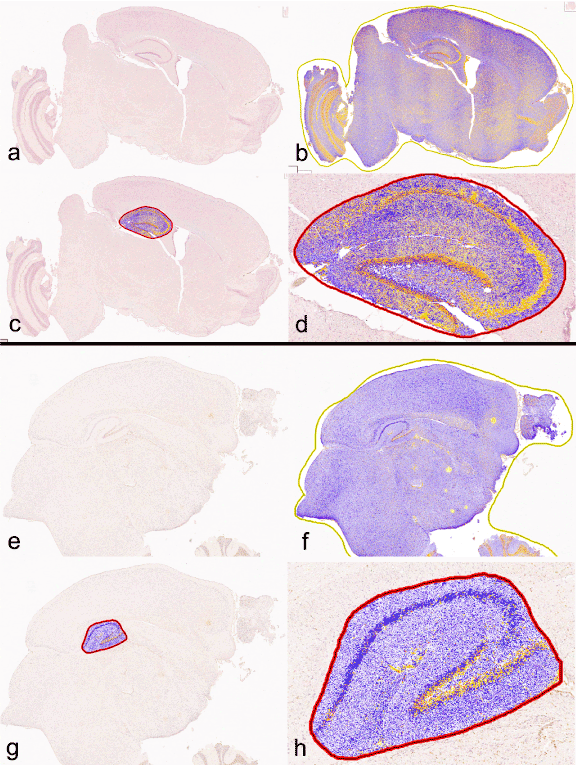
Figure 7. Example of Aperio Positive Pixel Count analysis using ImageScope. (a) Aβ mid-sagittal section. (b) Aβ mid-sagittal section segmented for DAB staining intensity. (c) Aβ mid-sagittal section with only hippocampus segmented. (d) magnified Aβ mid-sagittal section of segmented hippocampus. (e) HP-Tau mid-sagittal section. (f) HP-Tau mid-sagittal section segmented for DAB staining intensity. (g) HP-Tau mid-sagittal section with only hippocampus segmented. (h) magnified HP-Tau mid-sagittal section of segmented hippocampus. Color scale: Yellow = weak positive; Orange = positive; Red = strong positive; Purple/blue = negative. Only positive and strong positive pixels were included in the statistical analysis
Aβ Positive Pixel Count
Factorial ANOVA analysis revealed a significant main effect on whole slice deposition of Aβ for age [F(2,4) = 13.62, p = 0.02] and a trend toward significance for APOE genotype [F(2,4) = 6.18, p = 0.06].
Follow-up a posteriori analyses for age showed a significant decrease between 3-months and 6-months (p = 0.03) and 3-months and 12-months (p = 0.02). Analyses for APOE genotype showed a trend toward a significant increase between APOE2 and APOE3 (p = 0.07), and a significant decrease between APOE3 and APOE4 (p = 0.01).
Factorial ANOVA analysis revealed a significant main effect on hippocampal deposition of Aβ for age [F(2,4) = 6.80, p = 0.05] and a trend toward significance for APOE genotype [F(2,2)=4.62, p = 0.09).
Follow-up a posteriori analyses for age showed a trend toward a significant decrease between 3-months and 12-months (p = 0.08). Analyses for APOE genotype showed a significant decrease between APOE3 and APOE4 (p = 0.03).
HP-Tau Positive Pixel Count
Factorial ANOVA analysis revealed a significant main effect on whole slice deposition of HP-Tau for diet [F(1,13) = 10.90, p = 0.006].
Follow-up a posteriori analyses for diet showed a large, albeit non-significant decrease in HP-Tau deposition between control and L-cysteine diets without regard to APOE genotype (p = 0.18) (Figure 8).
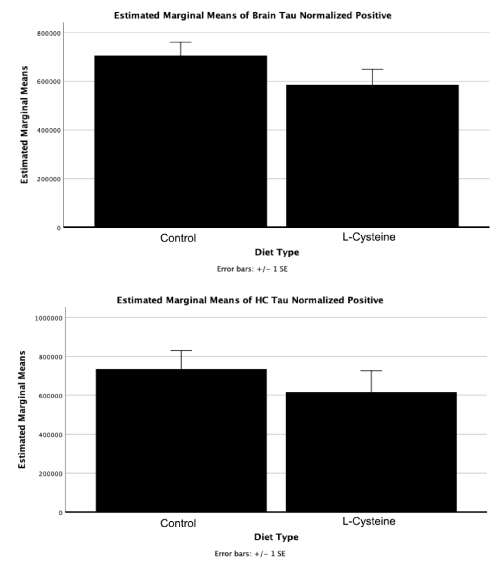
Figure 8. Positive pixel count decrease in HP-Tau staining intensity as a result L-Cysteine. Although the Factorial ANOVA main effect on whole slice deposition of HP-Tau was significant for diet [F(1,13) = 10.90, p = 0.006], pairwise comparisons between diet type were not (p = 0.18). Future experiments to increase statistical power are necessary to determine if this effect is statistically significant
Factorial ANOVA analysis revealed a significant main effect on whole slice deposition of HP-Tau for age [F(3,13) = 5.53, p = 0.01].
Follow-up a posteriori analyses for age showed a trend toward a significant increase between 6-months and 12-months (p = 0.06).
Effects of L-Cysteine in the mouse brain
L-cysteine had statistically significant effects on APOE abundance which differed among each cohort. For example, APOE3 abundance was decreased by L-cysteine, whereas APOE4 was increased; there was no effect seen with APOE2 (Figure 2). Although no statistically significant effects were observed concerning Aβ and HP-Tau deposition, there were several trends toward significance noted. For instance, we saw a trend toward a decrease in Aβ for the APOE3 cohort on the L-cysteine diet, despite seeing none with the APOE2 and APOE4 cohorts (Figure 1). These observations suggest that L-cysteine is sequestering iron in APOE3 brains, thus reducing the need for Aβ production and consequently, APOE3 production. Given that the decrease in APOE3 was statistically significant and the decrease in Aβ was not, it is likely that large amounts of APOE3 are produced in response to a relatively smaller Aβ burden. Interestingly, there was also a trend toward a decrease in HP-Tau deposition in the APOE3 cohort on the L-cysteine diet (Figure 6), which is similar to what we saw with Aβ, suggesting that lowering Aβ deposition consequently lowers HP-Tau deposition. Additionally, the fact that high levels of Aβ inhibit proteosomes that degrade HP-Tau is further evidence for this notion [13]. Another trend we saw was toward a decrease in HP-Tau deposition in the APOE2 cohort, but toward an increase in the APOE4 cohort on the L-cysteine diet (Figure 6). Thus, it is safe to conclude that the effect of L-cysteine on APOE2 individuals is mostly negligible, although somewhat positive, and is significantly beneficial for those with the APOE3 genotype.
However, the effect of L-cysteine on APOE4 individuals seems to be paradoxical. For instance, an increase in APOE4 abundance decreases Aβ deposition, since APOE binds and removes Aβ [2]; but an increase in HP-Tau deposition, because APOE4 inhibits the degradation of HP-Tau [11]. Conversely, a decrease in Aβ deposition naturally results in a decrease in HP-Tau deposition [13]. Our data show that the increase in APOE4 due to L-cysteine was statistically significant (Figure 2), that there was no change in Aβ deposition (Figure 4), and that there was merely a trend toward an increase in HP-Tau deposition for the APOE4 cohort (Figure 6). Moreover, it is clear from our Western blot that L-cysteine caused a decrease in APOE4 degradation (Figure 1). Given that the HtrA1 protein degrades APOE4 into 25-kD fragments in vitro [24], it is evident that L-cysteine is an inhibitor of HtrA1-mediated degradation of APOE4. HtrA1 also degrades Aβ and HP-Tau [25,26], which mechanisms may also be negatively affected by L-cysteine. The possible inhibition of HtrA1-mediated HP-Tau and Aβ degradation by L-cysteine would explain the trend toward an increase in HP-Tau deposition and the lack of an overall effect on Aβ in the APOE4 cohort. Thus, the effect of L-cysteine on APOE4 individuals remains well-balanced.
Positive pixel staining in the mid-sagittal brain and hippocampus sections
Immunohistochemistry analyses (Figure 7) revealed no effects of L-cysteine on Aβ, although a somewhat insignificant decrease in HP-Tau without regard to APOE genotype (p = 0.18) (Figure 8). Such results were expected, as we only used one brain section for immunohistochemistry analyses on Aβ and HP-Tau, but we used whole-hemispheres for Western blot analyses; whole hemisphere immunohistochemistry analysis might match Western blot results. Despite the lack of statistical significance, the results are interesting because they show that L-cysteine may cause a decrease in HP-Tau in the hippocampus and midsagittal brain (Figure 8). Immunohistochemistry testing of additional mice will reveal whether L-cysteine has any such effect. Furthermore, a heat map of co-localization to hippocampal cytoarchitectonic areas might be even more revealing. Thus, we plan to do these studies in the future to learn more about the effects of L-cysteine on AD in APOE2, APOE3, and APOE4 homozygous mice.
Possible mechanisms of treatment for L-Cysteine
As a treatment for AD, L-cysteine may act in a number of possible ways. For example, L-cysteine can sequester iron via its thiol, making iron-sulfur clusters and alanine (Figure 9) [27]. As a result, it is suggestive that Aβ will not be formed to sequester the iron, and thus Aβ plaque levels will decrease. Consequently, the inefficiency of APOE4 at clearing Aβ plaques would be negated.
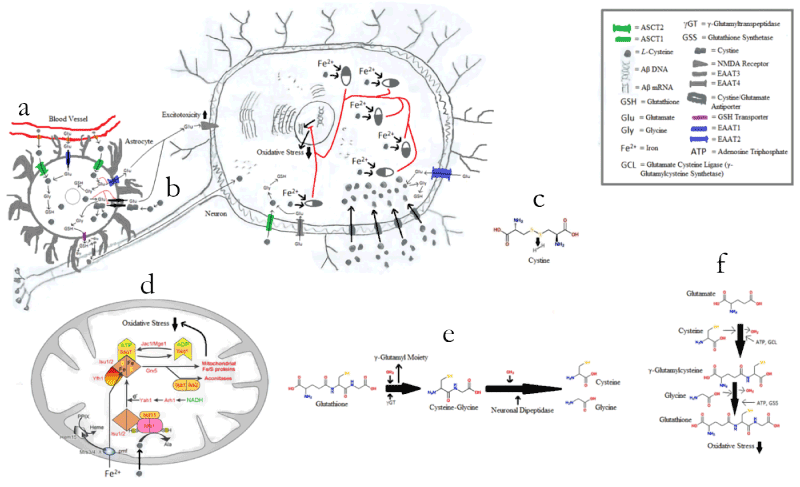
Figure 9. Possible Effects of L-Cysteine on Oxidative Stress. (a) L-cysteine combining with glutamate and glycine in an astrocyte to form GSH, and thus binding excess intracellular glutamate before that glutamate can be pumped out of the astrocyte via the cystine-glutamate antiporters or the broken EAAT2 transporters. (b) Free L-cysteine being oxidized in the body to form cystine, which works as a substrate for the cystine-glutamate antiporter. (c) Cystine synthesis from two free cysteines. (d) L-cysteine sequestering iron via its thiol, making iron-sulfur clusters and alanine (occurs inside the mitochondria). (e) GSH breakdown. (f) GSH synthesis
Additionally, free L-cysteine is oxidized in the body to form cystine (two L-cysteine molecules bound together via a disulfide bridge), which works as a substrate for the cystine-glutamate antiporter (Figure 9) [28]. The cystine-glutamate antiporter pumps cystine into the cell and glutamate out of the cell [28]. The cystine-glutamate antiporter is important in AD, because excitotoxicity due to excess glutamate release, as well as reduced uptake by impaired glutamate transporters, damages nerve cells and exacerbates AD via the binding of excess glutamate to the N-methyl-D-aspartate (NMDA) receptor [29-31]. So far, astrocytal excitatory amino acid transporter 2 (EAAT2) has been identified as an impaired glutamate transporter in AD [31], although the same discovery has not yet been made for neuronal EAAT2 [32]. EAAT2 is uncommon in neurons [32], but common in astrocytes [31], whereas EAAT3 is the most common neuronal transporter of glutamate and cysteine but is not found in astrocytes [33]. Moreover, glycine serves as a necessary co-agonist to glutamate for the NMDA receptor [29]. Interestingly, L-cysteine can combine with glutamate and glycine in cells to form the antioxidant GSH [34,35], and by so doing, potentially bind excess intracellular glutamate before that glutamate can be pumped out of the astrocytes via the cystine-glutamate antiporters or the broken EAAT2 transporters (Figure 9).
Furthermore, it is evident that L-cysteine is an inhibitor of HtrA1-mediated degradation of APOE4 (Figure 1), and possibly of Aβ and HP-Tau (Figures 4, 6). It is also clear that L-cysteine does not affect the ability of HtrA1 to degrade APOE3 (Figure 1). This finding is understandable, as HtrA1 degrades APOE3 to a significantly lesser extent than it degrades APOE4 [11]. However, the mechanism by which L-cysteine inhibits HtrA1-mediated degradation of APOE4, and potentially of Aβ and HP-Tau, remains elusive. It is plausible that free L-cysteine forms disulfide bonds with the abundance of cysteine residues concentrated in the N-terminal third of the HtrA1 molecule (Table 2), resulting in a conformational change non-conducive to its binding of APOE4, and possibly of Aβ and HP-Tau (Figure 10). More experiments are required to uncover this mechanism.
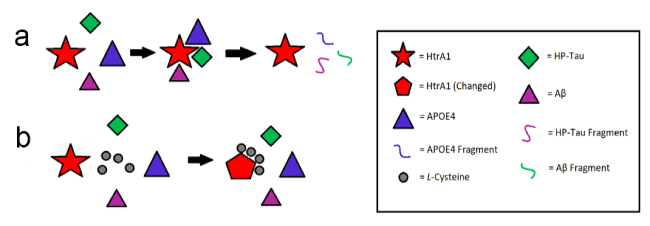
Figure 10. Possible Effect of L-Cysteine on HtrA1-Mediated Degradation of APOE4, Aβ, and HP-Tau. (a) No L-cysteine present. (b) L-cysteine present
Our effect was not as profound as what Chan, et al. (2008) described for the APOE4 cohort [1]. A possible explanation for this discrepancy is because NAC was used instead of L-cysteine [1]. NAC is a prodrug to L-cysteine created by attaching an acetyl group to enable the molecule to enter systemic circulation more easily after metabolism [36]. Therefore, a possible reason for the inefficiency of L-cysteine (relative to NAC) in relieving AD pathology could be because upon metabolism, too much is lost to have an effect on neurological function similar to NAC. Moreover, L-cysteine may only yield the degree of the effects seen by Chan, et al. (2008) when administered with ALCAR and/or SAMe [1]. On the other hand, L-cysteine may alleviate AD pathology to the same extent regardless of whether it is administered as NAC or in combination with ALCAR and/or SAMe. Additionally, the effect of L-cysteine on AD pathology in APOE4 homozygotes may be mostly due its ability to form GSH, a powerful antioxidant that naturally reduces oxidative stress [34,35]. Thus, further experiments are needed to determine the extent of the role of L-cysteine vs. NAC, as well as the roles of ALCAR and SAMe in relation to L-cysteine/NAC.
Although the results of Chan, et al. (2008) show much success, there remain problems with treatment application [1]. For instance, both NAC and SAMe have negative adverse effects in humans. Although NAC has not yet been proven to be carcinogenic, it is known to have cancer-exacerbating effects. For example, NAC increases proliferation of tumor cells by reducing ROS, DNA damage, and p53 expression in both mouse and human lung tumor cells [37-39]. High levels of ROS up-regulate p53 to induce apoptosis of damaged and/or cancerous cells [37-39]. Also, NAC has been reported to cause nausea, vomiting, fevers, and rashes when taken orally [40]. Additionally, SAMe has been reported to cause dyspepsia, gastrointestinal disorders, and anxiety [41]. SAMe is also a weak DNA-alkylating agent [42]. Taken together, this triple-combination diet appears to be potentially unsafe for long-term use in humans. On the other hand, there are no known adverse effects of L-cysteine and ALCAR. L-cysteine is converted to alanine after donating its thiol to sequester iron [27] and an increase in alanine levels leads to an increase in alanine aminotransferase (ALT) levels, which in turn increases the risk of developing type 2 diabetes mellitus [43]. However, the increase in alanine caused by the sequestering of iron by L-cysteine would likely not be enough to raise ALT to dangerous levels.
Although we achieved statistical significance on a major parameter using the mice we had, with so many statistical factors to consider, we were underpowered. For example, we had only 21 mice to run immunohistochemistry tests for Aβ and only 39 mice for HP-Tau. Moreover, due to technical difficulties with staining, we were not able to perform immunohistochemistry for the presence of APOE. Future studies will include more mice per experimental condition as well as an immunohistochemistry analysis of APOE abundance.
The authors wish to thank the following funding sources: Brigham Young University, College of Life Sciences, Mentoring Environment Grant; Brigham Young University, School of Family Life, Gerontology Program; Brigham Young University, Magnetic Resonance Imaging Research Facility, Seed Grant; Dr. Sarah M. McGinty, Neuroscience Graduate Student Research Fellowship; Neurodar, LLC; Limitless Worldwide, LLC.
- Shea TB (2007) Effects of dietary supplementation with N-acetyl cysteine, acetyl-L-carnitine and S-adenosyl methionine on cognitive performance and aggression in normal mice and mice expressing human ApoE4. Neuromolecular Med 9: 264-269.
- Rolyan H, Feike AC, Upadhaya AR, Waha A, Van Dooren T, et al. (2011) Amyloid-β protein modulates the perivascular clearance of neuronal apolipoprotein E in mouse models of Alzheimer's disease. J Neural Transm (Vienna) 118: 699-712. [Crossref]
- Espiritu DJ, Mazzone T (2008) Oxidative stress regulates adipocyte apolipoprotein e and suppresses its expression in obesity. Diabetes 57: 2992-2998.
- Bousejra-ElGarah F, Bijani C, Coppel Y, Faller P, Hureau C (2011) Iron (II) binding to amyloid-β, the Alzheimer’s peptide. Inorganic chemistry 50: 9024-9030.
- Ghebranious N, Ivacic L, Mallum J, Dokken C (2005) Detection of ApoE E2, E3 and E4 alleles using MALDI-TOF mass spectrometry and the homogeneous mass-extend technology. Nucleic Acids Res 33: e149.
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, et al. (1994) Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nature genetics 7: 180-184.
- Razali R, Zanariah MS, Elinda T, Wan Zurinah W, Mian TS, et al. (2013) Apolipoprotein e genotypes and behavioural and psychological symptoms of dementia (bpsd) in malaysian patients with dementia. Sains Malaysiana 42: 409-416.
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, et al. (2011) Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med 3: 89.
- Aleshkov SB, Li X, Lavrentiadou SN, Zannis VI (1999) Contribution of Cysteine 158, the Glycosylation Site Theonine 194, the Amino-and Carboxy-Terminal Domains of Apolipoprotein E in the Binding to Amyloid Peptide β (1-40). Biochemistry 38: 8918-8925.
- Jofre‐Monseny L, Minihane AM, Rimbach G (2008) Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol Nutr Food Res 52: 131-145.
- Chu Q, Diedrich JK, Vaughan JM, Donaldson CJ, et al. (2016) HtrA1 Proteolysis of ApoE In Vitro Is Allele Selective. J Am Chem Soc 138: 9473-9478. [Crossref]
- Mondragón-Rodríguez S, Perry G, Zhu X, Moreira PI, Acevedo-Aquino MC, et al. (2013) Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: implications for Alzheimer’s disease. Oxid Med Cell Longev 2013: 940603.
- Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM (2008) Aβ inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging 29: 1607-1618.
- Aleshkov S, Abraham CR, Zannis VI (1997) Interaction of Nascent ApoE2, ApoE3, and ApoE4 Isoforms Expressed in Mammalian Cells with Amyloid Peptide β (1− 40). Relevance to Alzheimer's Disease. Biochemistry 36: 10571-10580.
- Hanson AJ, Bayer-Carter JL, Green PS, Montine TJ, Wilkinson CW, et al. (2013) Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA Neurol 70: 972-980.
- Wernette-Hammond ME, Lauer SJ, Corsini A, Walker D, Taylor JM, et al. (1989) Glycosylation of human apolipoprotein E. The carbohydrate attachment site is threonine 194. J Biol Chem 264: 9094-9101.
- Ioannou YA, Zeidner KM, Grace ME, Desnick RJ (1998) Human alpha-galactosidase A: glycosylation site 3 is essential for enzyme solubility. Biochem J 332: 789–797.
- Tams JW, Vind J, Welinder KG (1999) Adapting protein solubility by glycosylation.: N-Glycosylation mutants of Coprinus cinereus peroxidase in salt and organic solutions. Biochim Biophys Acta 1432: 214-221.
- Høiberg-Nielsen R, Fuglsang CC, Arleth L, Westh P (2006) Interrelationships of glycosylation and aggregation kinetics for Peniophora lycii phytase. Biochemistry 45: 5057-5066.
- Lee Y, Kockx M, Raftery MJ, Jessup W, Griffith R, et al. (2010) Glycosylation and sialylation of macrophage-derived human apolipoprotein E analyzed by SDS-PAGE and mass spectrometry: evidence for a novel site of glycosylation on Ser290. Molecular & Cellular Proteomics 9: 1968-1981.
- Abrams AJ, Farooq A, Wang G (2011) S-nitrosylation of ApoE in Alzheimer's disease. Biochemistry 50: 3405-3407. [Crossref]
- Marmillot P, Rao MN, Liu QH, Lakshman MR (1999) Desialylation of human apolipoprotein E decreases its binding to human high-density lipoprotein and its ability to deliver esterified cholesterol to the liver. Metabolism 48: 1184-1192.
- LaDu MJ, Munson GW, Jungbauer L, Getz GS, Reardon CA, et al. (2012) Preferential interactions between APOE-containing lipoproteins and Abeta revealed by a detection method that combines size exclusion chromatography with non-reducing gel-shift. Biochim Biophys Acta - Molecular and Cell Biology of Lipids 1821: 295-302.
- Muñoz SS, Li H, Ruberu K, Chu Q, Saghatelian A, et al. (2018) The serine protease HtrA1 contributes to the formation of an extracellular 25-kDa apolipoprotein E fragment that stimulates neuritogenesis. J Biol Chem 293: 4071-4084.
- Grau S, Baldi A, Bussani R, Tian X, Stefanescu R, et al. (2005) Implications of the serine protease HtrA1 in amyloid precursor protein processing. Proc Natl Acad Sci USA 102: 6021–6026.
- Tennstaedt A, Pöpsel S, Truebestein L, Hauske P, Brockmann A, et al. (2012) Human High Temperature Requirement Serine Protease A1 (HTRA1) Degrades Tau Protein Aggregates. J Biol Chem 287: 20931–20941.
- Lill R, Mühlenhoff U (2006) Iron-sulfur protein biogenesis in eukaryotes: components and mechanisms. Annu Rev Cell Dev Biol 22: 457-486. [Crossref]
- Lo M, Wang YZ, Gout PW (2008) The x cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J Cell Physiol 215: 593-602.
- Liu Y, Zhang J (2000) Recent development in NMDA receptors. Chin Med J (Engl) 113: 948-956. [Crossref]
- Hynd MR, Scott HL, Dodd PR (2004) Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem Int 45: 583-595.
- Yi JH, Hazell AS (2006) Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int 48: 394-403.
- Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, et al. (2008) A Quantitative Assessment of Glutamate Uptake into Hippocampal Synaptic Terminals and Astrocytes: New Insights into a Neuronal Role for Excitatory Amino Acid Transporter 2 (EAAT2). Neuroscience 157: 80-94.
- Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, et al. (2012) The Density of EAAC1 (EAAT3) Glutamate Transporters Expressed by Neurons in the Mammalian CNS. J Neurosci 32: 6000-6013.
- Dringen R, Pfeiffer B, Hamprecht B (1999) Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci 19: 562-569.
- Aoyama K, Watabe M, Nakaki T (2008) Regulation of neuronal glutathione synthesis. J Pharmacol Sci 108: 227-238.
- Tirouvanziam R, Conrad CK, Bottiglieri T, Herzenberg LA, Moss RB, et al. (2006) High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc Natl Acad Sci USA 103: 4628-4633.
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, et al. (2000) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 97: 2892–2897.
- Bensaad K, Vousden KH (2005) Savior and slayer: the two faces of p53. Nat Med 11: 1278-1279. [Crossref]
- Chen W, Jiang T, Wang H, Tao S, Lau A, et al. (2012) Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid Redox Signal 17: 1670–1675.
- DailyMed (2014) Label: ACETYLCYSTEINE- Acetylcysteine inhalant. U.S. National Library of Medicine.
- Najm WI, Reinsch S, Hoehler F, Tobis JS, Harvey PW (2004) S-Adenosyl methionine (SAMe) versus celecoxib for the treatment of osteoarthritis symptoms: A double-blind cross-over trial. BMC Musculoskelet Disord 5: 6.
- Rydberg B, Lindahl T (1982) Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J 1.2: 211–216.
- Sattar N, Scherbakova O, Ford I, O’Reilly DS, Stanley A, et al. (2004) Elevated alanine aminotransferase predicts new-onset type 2 diabetes independently of classical risk factors, metabolic syndrome, and C-reactive protein in the west of Scotland coronary prevention study. Diabetes 53: 2855-2860.