Osteoclastoma is an infrequent, benign, primary bone neoplasm initially scripted by Jaffe in 1940 [1]. Osteoclastoma is an osteolytic, locally aggressive tumefaction incriminating young adults with skeletal maturity and is accompanied by an unpredictable biological course. Osteoclastoma is constituted by mononuclear, spheroid to spindle-shaped cells intermingled with numerous, evenly distributed, osteoclast- like, multinucleated giant cells [1,2].
Osteoclastoma, additionally nomenclated as giant cell tumour of bone or myeloid sarcoma, usually arises within metaphysis and extends into epiphysis of long bones. Non epiphyseal osteoclastoma is extremely exceptional. The neoplasm is rarely discerned within immature skeleton [2].
An exceptional neoplasm, osteoclastoma constitutes an estimated 3% to 5% of primary bone tumours. Osteoclastoma predominantly incriminates young adults amidst 20 years to 30 years with a peak incidence betwixt 3rd to 4th decade. The tumour is uncommonly discerned beneath <20 years or beyond >55 years. An estimated 10% instances appear within 15 years to 20 years [2,3]. Although osteoclastoma is benign, the tumefaction can infrequently undergo malignant metamorphoses in an estimated <2% subjects, commonly betwixt 30 years to 50 years. Osteoclastoma can exceptionally metastasize to pulmonary parenchyma. A slight female preponderance is observed [2].
Mononuclear cells constituting an osteoclastoma are derived from primitive mesenchymal cells. Usually osteoclastoma is discerned within long bones of extremities whereas sacrum or vertebral bodies are rarely involved. Osteoclastoma is commonly detected within epiphysis and adjacent metaphysis of long bones such as distal femur, proximal tibia, distal radius or proximal humerus. Adjunctive, incriminated bones are proximal sacrum, vertebrae or base of skull although no site of tumour emergence is exempt. Osteoclastoma is infrequently discerned within craniofacial bones, vertebral bodies, sacrum or small bones of hands and feet. An estimated 3% to 4% of osteoclastomas are situated within small bones of hands and feet [2,3].
Majority (>95%) of osteoclastomas are uni-focal. Osteoclastoma is predominantly a solitary neoplasm, appears in absence of concurrent Paget’s disease of bone and commonly incriminates distal femur and proximal tibia in around 50% to 65% instances besides implicating distal radius, sacrum or vertebral bodies. Multi-centric osteoclastoma is discerned in below <1% instances and arises in younger subjects prior to epiphyseal closure [3].
Prevalence of osteoclastoma is enhanced in Paget’s disease of bone with typical incrimination of pelvic or skull bones. Multifocal osteoclastoma in the absence of pre-existing Paget’s disease is exceptional. Osteoclastoma can also occur in concurrence with focal dermal hypoplasia (Goltz syndrome) [3,4].
Osteoclastoma can engender pathological fracture with thinning of bony cortex, penetrate the cortex and extend into adjacent soft tissues. Pathological fractures are observed in approximately 5% to 12% subjects. Osteoclastoma of small bones delineates enhanced tumour reoccurrence, in contrast to tumefaction arising in long bones. As osteoclastoma is uncommonly detected within bones of hands or feet, tumefaction observed within aforesaid sites is possibly a giant cell lesion of small bones whereas jaw is probably involved by a central giant cell granuloma [3,4].
Pathophysiology of osteoclastoma is inadequately exemplified. Mesenchymal cells appear as osteoblastic progenitors and enunciate receptor activator of nuclear factor kappa-B ligand (RANKL).
Receptor activator of nuclear factor kappa-B and its ligand (RANK/ RANKL) signalling pathway is extensively exemplified within osteoblast - like mononuclear stromal cells with consequent transformation of pre- osteoclast cells of monocytic lineage into osteoclast cells. Aforesaid osteoclasts initiate bone absorption with consequent osteolysis discerned within osteoclastoma [4,5].
Giant cells are non-neoplastic. Neoplastic cells are primitive, mononuclear stromal cells of mesenchymal origin which are constituted by pre-osteoblasts. Stromal cells manifest RANKL and pre-osteoblast markers such as alkaline phosphatase, runt- related transcription factor 2 (RUNX2), transcription factor Sp7 (osterix) and osteoclastin. RANKL molecules appearing on neoplastic pre-osteoblasts adhere to RANK (receptor activator of nuclear activator kappa-B) situated upon non-neoplastic, macrophage- like, osteoclast precursors, in concurrence with macrophage colony stimulating factor (M-CSF). Aforesaid combination promotes differentiation of osteoclasts with consequent giant cell formation [4,5].
Osteoclastoma commonly implicates epiphyses and adjacent metaphysis of long bones. Clinically, a lytic lesion within epiphyseal region of bone is commonly discerned which denominates as an epiphyseal, well circumscribed, lytic neoplasm detected on plain radiography. Osteoclastoma is a neoplasm of obscure aetiology and unpredictable biological behaviour. Thus, cogent histological assessment is necessary for confirmation of the neoplasm [5].
Osteoclastoma commonly manifests joint pain, swelling and restricted mobility on account of tumour proximity to joint space. Pain is persistent, deep-seated and unrelated to trauma to the joint. Knee joint frequently displays aforesaid symptoms as epiphysis of distal femur or proximal tibia is a frequent (>50%) site of tumour incrimination [5]. Osteoclastoma is asymptomatic or discerned incidentally or can initially represent as a pathological fracture or infrequently as pulmonary metastatic disease [5].
Malignant transformation is exceptional and can be primary or secondary. Typically, primary malignancy occurs adjacent to benign foci of osteoclastoma whereas secondary malignancy arises upon site of preceding treatment, generally radiotherapy [5,6].
Primary, malignant osteoclastoma is a high-grade sarcoma uncommonly emerging within an osteoclastoma upon initial discernment. Secondary, malignant osteoclastoma is a commonly discerned, high grade sarcoma pre-eminently arising within therapy induced site of a benign osteoclastoma, following surgery or employment of low dose radiation therapy. Malignant osteoclastoma is associated with an inferior survival [5,6]. Malignant osteoclastoma is extremely exceptional and appears within older age group, as compared to benign osteoclastoma. Sites incriminated are identical and the neoplasm appears in concurrence with pre-existing osteoclastoma or radiation therapy. Appropriate ascertainment of secondary, malignant osteoclastoma mandates delineation of pre-existing or co-existing benign osteoclastoma within incriminated location of emergent spindle cell sarcoma [5,6].
Although several classifications are proposed for staging osteoclastoma, Campanacci grading system is commonly adopted as tumour categorization is contingent to radiological and clinical appearance [2,3].
Grade I - an intraosseous, quiescent lesion confined to medullary canal with an absence of cortical incrimination, well circumscribed tumour margin and an intact bony perimeter.
Grade II –expansible, active, intraosseous lesion with well-defined tumour perimeter along with thinned-out, expanded, contiguous cortex.
Grade III- extra-osseous, aggressive, radiolucent lesion with ill-defined tumour perimeter, cortical discontinuity and tumour extension into adjacent soft tissue [2,3].
However, aforesaid classification fails to predict localized tumour reoccurrence or tumour metastasis [3].
Grossly, osteoclastoma demonstrates a well-defined lesion perimeter. The tumefaction is expansible with cortical thinning, soft tissue extension and pushing tumour margin, incompletely rimmed with reactive bone [5].
Cut surface exhibits yellow, xanthomatous, whitish, fibrous, haemorrhagic or cystic areas. Foci of necrosis are encountered in enlarged tumefaction. Malignant metamorphosis represents as enlarged, fleshy areas with infiltration of circumscribing soft tissue [5,6].
Osteoclastoma is a predominantly osteoclastogenic, stromal neoplasm, composed of mononuclear cells, macrophages and innumerable, osteoclast-like, multinucleated giant cells uniformly distributed throughout the tumefaction.
Giant cells do not circumscribe haemorrhagic areas, in contrast to central giant cell granuloma, giant cell lesion of small bones or aneurysmal bone cyst [5,6]. Osteoclastoma demonstrates foci of evenly disseminated, elongated, elliptical or polygonal mononuclear cells. Giant cells constituting osteoclastoma are reactive osteoclasts. Mononuclear stromal cells configure the neoplastic and proliferative component of osteoclastoma. Also, neoplastic stromal cells can competently induce osteoclast-like cellular differentiation [5,6]. Mononuclear monocytes are contemplated as osteoclast precursor cells. Atypical mitosis or marked cytological atypia is absent although mononuclear stromal cells can infrequently depict mitotic figures. Mammoth giant cells can exceed the magnitude of normal osteoclasts and display several nuclei, usually exceeding > 50. Also, mitotic activity is absent in multinucleated giant cells. Focal paucity of giant cells is observed [5,6].
Spheroidal, spindle-shaped or elliptical mononuclear cells are an essential component. Macrophage- like and primitive mesenchymal cells with ill-defined cytoplasm and mitotic activity articulate the neoplastic element of osteoclastoma. Intervening stroma is extensively vascular and commonly depicts foci or reactive, woven bone and fibrosis. Osteoclastoma exhibits focal acute haemorrhage, haemosiderin pigment deposits, accumulated xanthomatous histiocytes and zonal necrosis within enlarged tumours. Alterations akin to secondary aneurysmal bone cyst are encountered in an estimated 10% instances. Vascular infiltration can be detected within tumour perimeter [6,7]. Pulmonary metastasis can ensue although the feature is insignificant vis-à-vis prognostic outcomes. Malignant osteoclastoma morphologically simulates high grade sarcoma such as osteosarcoma, fibrosarcoma or malignant fibrous histiocytoma. The characteristic, well defined, histological configuration of osteoclastoma is frequently deranged by secondary, reactive modifications incriminating mononuclear stromal cells such as fibro-histiocytic cellular proliferation, haemorrhage, necrosis or genesis of aneurysmal bone cyst [5,7]. Malignant cytological features ensue with malignant metamorphoses. Pulmonary metastasis displays an identical histology as the primary neoplasm and is associated with an excellent prognosis. Albeit, localized tumour aggression or occurrence of pulmonary metastasis is lacks concurrence with histological grading (Figures 1-8) [6-16].
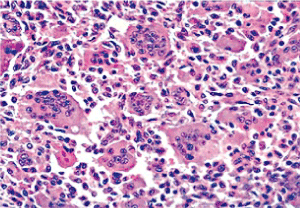
Figure 1. Osteoclastoma delineating regularly distributed multinucleated giant cells intermixed with a stroma of mononuclear cells and foci of fibro-histiocytic proliferation [9]
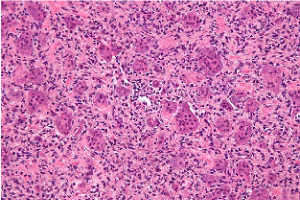
Figure 2. Osteoclastoma exhibiting multinucleated giant cells evenly disseminated betwixt a stroma of mononuclear cells and fascicles of fibro-histiocytic proliferation [10]
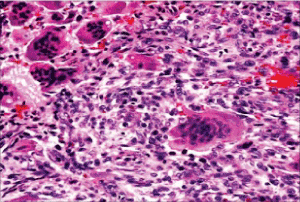
Figure 3. Osteoclastoma with even dispersal of multinucleated giant cells scattered amidst a stroma of mononuclear cells, red cell exudation and fibro-histiocytic proliferation [11]
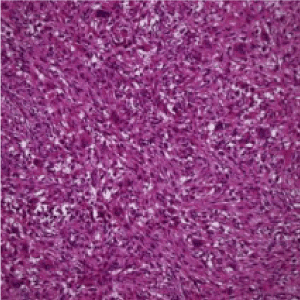
Figure 4. Osteoclastoma comprised of uniform, multinucleated giant cells disseminated within a stroma rich in mononuclear cells of monocytic lineage and foci of fibro-histiocytic proliferation with red cell extravasation [12]
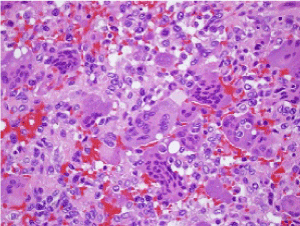
Figure 5. Osteoclastoma exemplifying focal haemorrhage, multinucleated giant cells and scattered, mononuclear stromal cells [13]
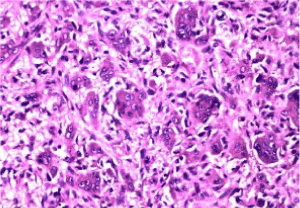
Figure 6. Osteoclastoma depicting evenly disseminated multinucleated giant cells, stromal clusters of mononuclear cells and foci of fibro-histocytic proliferation, lack of haemorrhage or mitosis [14]
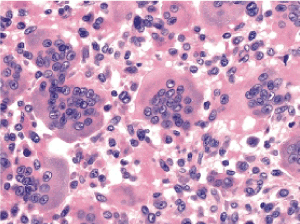
Figure 7. Osteoclastoma delineating regularly dispersed multinucleated giant cells, mononuclear cells within the stroma and fibro-histocytic proliferation [15]
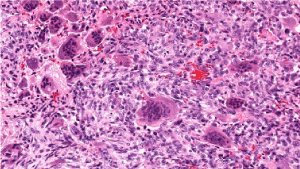
Figure 8. Osteoclastoma demonstrating evenly distributed multinucleated giant cells intermingled with mononuclear cells of monocytic lineage and foci of haemorrhage [16]
Giant cells delineate immune phenotype of osetoclasts and are immune reactive to CD45, CD68 and cathepsin K. Immune staining for p63 is discerned in a majority (>95%) of instances although mononuclear cells depict an immune staining of variable intensity. Nevertheless, immune staining with p63 is non-specific for osteoclastoma and can also be discerned within adjunctive giant cell –rich, osteotic lesions [3,4].
As appropriate histological discernment of osteoclastoma is challenging, chromosomal mutation of H3F3A gene, delineated in an estimated 95% to 100% of osteoclastomas of long bones is pertinent in differentiating the neoplasm from adjunctive entities. Thus, monoclonal antibody indicating G34W site of chromosomal mutation within H3F3A gene can be employed for suitable immune histochemical enunciation of the neoplasm [3,4].
Antihistone H3.3 G34W antibody is immune reactive wherein nuclear staining is detected in a majority (>90%) mononuclear cells. On cytogenetic analysis, Histone H3F3A (H3.3) G34W mutation is exemplified in a majority (96%) of subjects [3,4].
Osteoclastoma mandates a segregation from conditions such as fibrous metaphyseal defect, giant cell- rich osteosarcoma, metastatic carcinoma, chondroblastoma, chondromyxoid fibroma, non-ossifying fibroma, aneurysmal bone cyst, endochondroma, chondrosarcoma and desmoplastic fibroma [5,6].
Osteoclastoma requires a distinction from brown tumour of hyperparathyroidism, a neoplasm which occurs in concurrence with hyperparathyroidism. The multifocal neoplasm is morphologically identical to osteoclastoma of small bones or central giant cell granuloma. Central giant cell granuloma microscopically recapitulates aneurysmal bone cyst or osteoclastoma of small bones although emerges within the jaw. Albeit, giant cells are unevenly dispersed within the stroma [5,6]. Chondroblastoma is a neoplasm which arises within epiphysis, akin to osteoclastoma. Tumour is comprised of pavement- like, cellular aggregates of uniform, spheroidal to polygonal cells with well-defined cell membranes, cellular nuclei with longitudinal grooves and peri-cellular “chicken wire” calcification along with a smattering of giant cells within the neoplasm [5,6].
Giant cell lesion of small bones commonly incriminates small bones of hands and feet. The site is uncommon for emergence of an osteoclastoma. Dissemination of giant cells is disorderly, and cells are aggregated around haemorrhagic foci, as exemplified with aneurysmal bone cyst [5,6].
As fibro- histiocytic proliferative reaction is prominent, osteoclastoma mandates a differentiation from non-ossifying fibroma (NOF) and benign fibrous histocytoma. Non ossifying fibroma predominantly occurs within bone metaphysis, stromal component is markedly fibroblastic, configures a storiform pattern and giant cells dissemination is non uniform. Non ossifying fibroma exemplifies a culmination of reparative process within the neoplasm following specific, secondary phenomenon such as haemorrhage, necrosis, lipoidization, scarring or fibro-histiocytic proliferation [5,6].
Non ossifying fibroma (NOF) is a neoplasm confined to immature skeleton. Microscopically, bland, spindle-shaped cells accompanied by fibroblasts articulate miniature fascicles to configure a storiform pattern along with scattered, osteoclast- like giant cells. However, giant cells are not a significant feature [5,6].
Primary aneurysmal bone cyst (ABC) demonstrates fluid- filled levels on plain radiography. Morphologically, giant cells are unevenly dispersed and are accumulated around blood- filled cystic spaces. Also, fibrous tissue septa comprised of bland, spindle-shaped cells, fibroblasts, myofibroblasts and fragments of woven bone are discerned, traversing the neoplasm. Genomic rearrangements of USP6 gene are discerned in an estimated 70% instances, a feature which is absent in osteoclastoma. Miniature, irregularly dispersed giant cells are commonly articulated within an aneurysmal bone cyst. Solid foci of aneurysmal bone cyst are fibrotic. Aneurysmal bone cyst- like solid tumour areas are commonly discerned in osteoclastoma [5,6].
Tumour necrosis in association with or absence of haemorrhagic foci are occasionally discerned in conventional osteoclastoma. Mononuclear stromal cells abutting necrotic areas can depict focal cytological atypia, akin to malignant metamorphoses. Atypical mitosis is absent, thus indicating the benign nature of neoplasm.
Tenosynovial giant cell tumour is a soft tissue neoplasm demonstrating sparse or abundant giant cells. Bony incrimination may be absent. Benign fibrous histiocytoma delineates an absence of broad expanse of mononuclear cells [5,6]. Giant cell- rich osteosarcoma requires a distinction from osteoclastoma.
Giant cell -rich osteosarcoma enunciates significant nuclear pleomorphism, atypical mitotic figures and characteristic configuration of malignant osteoid.
Osteoclastoma of small bones necessitates a separation from giant cell reparative granuloma. An absence of mononuclear stromal cells is encountered in giant cell reparative granuloma [5,6].
Abnormal biochemical parameters are discerned in osteoclastoma as observed with elevated alkaline phosphate levels. Although associated with aggressive features on imaging, osteoclastoma is a low-grade neoplasm [7].
Establishing a diagnosis of osteoclastoma necessitates adequate tissue evaluation wherein suspicious lesions require ascertainment of malignant metamorphoses. Procurement of ultrasound or computerized tomography (CT) guided tissue specimens are appropriate for tumour assessment with pertinent radiographic investigations [7]. Osteoclastoma can be suitably discerned with the employment of several imaging modalities such as plain radiographs, computerized tomography (CT), magnetic resonance imaging (MRI) or the employment of nuclear medicine [7].
Plain radiographic evaluation of primary tumour site is preferred. Computerized tomography (CT) can also be adopted. Computerized tomographic (CT) scan or plain radiographs of thoracic cavity are also recommended to detect pulmonary metastasis of osteoclastoma. Multi-centric osteoblastoma mandates evaluation with radionuclide bone scan [7,8].
On plain radiography, a lobulated, expansible, osteolytic lesion with a classic “soap bubble” appearance is exemplified. The neoplasm can extend within circumscribing soft tissues and delineate an “eggshell” ossification upon tumour periphery [7]. Osteoclastoma of long bones display cogent radiographic findings such as an eccentric lesion with well defined, non-sclerotic border, tumour appearing within closed growth plates and abutment of an articular surface. However, as osteoclastoma displays a penchant for pulmonary metastasis, national comprehensive cancer network (NCCN) recommends the employment of computerized tomography (CT) of thoracic cavity [7,8].
On CT scan, osteoclastoma appears as an eccentric, lytic lesion with well defined, non-sclerotic bony perimeter, confined to the metaphysis. Magnetic resonance imaging (MRI) demonstrates low to intermediate signal intensity upon T1 weighted imaging and a heterogeneous, elevated signal intensity upon T2 weighted imaging with signal enhancement [7,8]. Nuclear assay with technetium 99 scintigraphy exhibits elevated radiotracer upon delayed imaging with a peripheral prominence and a photopenic centroidal region [8].
Osteoclastoma demonstrates an overall favourable prognosis although percentage of localized tumour reoccurrence is variable. Prognostic outcomes are contingent to extent of surgical resection wherein localized tumour excision with a broad perimeter of normal tissue is associated with minimal tumour reoccurrence although enhanced morbidity is detected, in contrast to adoption of adjuvant therapy. Osteoclastoma abutting the spinal cord delineates an unfavourable prognosis with enhanced localized tumour reoccurrence [8].
Tumour relapse is up to an estimated 50% with treatment manoeuvers such as intra- lesional curettage in the absence of concurrent adjuvant therapy and around 75% within two years. Thus, to reduce tumour reappearance local adjuvant therapy can be suitably combined with surgical eradication of the tumefaction. Decimated tumour reoccurrence appears at roughly 13% to 22% [7,8].
An estimated 50% instances depict tumour reoccurrence following curettage within 3 years. Exceptionally, tumour reoccurrence can arise after three years. Adoption of localized tumour excision with broad perimeter of normal tissue can decimate tumour relapse. Pulmonary metastasis of osteoclastoma recapitulates morphology of the parent tumour. Metastasis are gradually progressive, non-aggressive and can spontaneously retrogress. Localized tumour reoccurrence, high tumour grade on Campanacci scale and intra-lesional curettage are possible factors for initiating pulmonary metastasis. Pulmonary tumour nodules are amenable to surgical extermination and display a superior therapeutic outcome [7,8].
Infrequently discerned malignant metamorphosis of osteoclastoma exhibits an inferior prognosis. Secondary malignant osteclastoma emerging on account of therapeutic intervention of benign osteoclastoma demonstrates a significantly unfavourable outcome, akin to adjunctive high-grade sarcoma, in contrast to primary malignant osteoclastoma [7,8].
Recommended surveillance following therapy typically includes CT imaging of primary site at three months following treatment, twice a year over the next two years and subsequently once a year for a duration of five years [8].
Osteoclastoma is adequately managed with tumour curettage or appropriate surgical resection of the neoplasm with broad perimeter of uninvolved tissue. Aforesaid therapeutic measures typically depict a favourable prognosis.
Intra-lesional tumour curettage is a suitable option for treating osteoclastoma. However, adoption of a singular procedure is accompanied by around 15% to 50% tumour reoccurrence within 2 years [7,8].
Preferred treatment of osteoclastoma incriminating the appendicular skeleton is surgical extermination of the lesion. Surgical manoeuver adopted is contingent to tumour magnitude, tumour localization and occurrence of concomitant pathological fracture. Cogent surgical options are intra-lesional curettage in concurrence with or absence of bone cement, en bloc tumour excision or marginal resection of the tumefaction with removal of a perimeter of normal, uninvolved tissue [7,8]. Occasionally, extensive surgical eradication is required, especially in instances of significantly damaged articular surface, repetitive tumour reoccurrence, extra-osseous tumour extension or dislocated, pathological fracture [7,8].
Osteoclastoma can be appropriately managed with a combination of surgery, radiotherapy and monoclonal antibody denosumab. Surgical tumour resection is a primary, preferential therapeutic option. Anti-osteoclast agents such as bisphosphonates or anti-RANKL monoclonal antibody denosumab can be efficaciously employed. Administration of denosumab with concomitant computerized tomography (CT) of thoracic cavity followed by chest and whole lung irradiation can be adopted. Radiation therapy or denosumab are efficacious for treating candidates unamenable to surgery or in instances with enhanced surgical morbidity [7,8]. Denosumab is a monoclonal antibody which effectively adheres to RANKL and impedes the activation receptor RANK. Consequently, functional inhibition and recruitment of osteoclast- like cells is denominated. Osteoclastoma treated with densoumab demonstrates manifestations such as ossification, fibrosis, reduction of giant cells, proliferation of mononuclear cells and cytological atypia [7,8]. Denosumab is employed wherein
a) significant morbidity is expected or incurred from initial surgical manoeuver
b) Osteoclastoma associated with localized tumour reoccurrence
c) candidates who are poor surgical risks
d) treatment of tumours situated within inaccessible locations. Proportionate tumour reoccurrence and significant morbidity accompanies preliminary surgical intervention [7,8].
Traditionally, intra-lesional curettage along with impaction of bone cavity with cortico-cancellous bone chips is adopted. Contemporary treatment methods such as bone cementing with poly methyl methacrylate (PMMA) is promising as it aids bone reconstruction and reduces localized tumour reoccurrence. Incurrence of an exothermic reaction with consequent localized hyperthermia can induce necrosis of tumour tissue remnants. As PMMA is commonly applied adjacent to articular cartilage and subchondral bone, degenerative arthritis can ensue. Radiolucent zone develops amidst bone- cement interface with eventual progression into foci of osteolysis [7,8]. Interferon alpha can also be utilized to treat osteoclastoma.
Alternatively, arterial embolization can be adopted. Bisphosphonates can also be beneficially administered. Therapeutic measures can be complemented with cogent radiotherapy [8]. Pulmonary metastasis is minimally aggressive, in contrast to solid osteoclastoma. Tumour reappearance with surgical therapy is up to an estimated 50%. Localized therapeutic adjuvants such as aqueous zinc chloride, PMMA, phenol, argon beam laser coagulation or cryotherapy can be utilized to decimate tumour relapse. PMMA is a preferential therapeutic adjuvant as it is associated with significant decline in tumour reoccurrence. Also, application of phenol is associated with chemical burns [7,8]. Localized tumour reoccurrence can be suitably treated with additional curettage of the neoplasm in combination with localized adjuvant therapy. Aforesaid treatment methodology is accompanied with minimally enhanced tumour morbidity. Approach to spinal neoplasms for a comprehensive surgical eradication is challenging as is extermination of sacral tumours due to proximity of sacral nerves [7,8]. Therapeutic outcomes are focussed at minimizing individual morbidity and maximizing appropriate function of affected bone. Surgical extermination of osteoclastoma is accompanied by several complications, significant morbidity and deterioration of visceral functions. Ingestion of denosumab can engender osteonecrosis of the jaw. Exposure to radiation enhances probable malignant metamorphoses [7,8].
- Jaffe HL, Lichtenstein L, Portis RB (Giant cell tumour of bone -It’s pathologic appearance, grading, supposed variants and treatment. Arch Pathol 30: 993-1031.
- Kuhn J, Pathak S (2020) Osteoclastoma. Stat Pearls Publishing 2020 Treasure Island Florida.
- Mavrogenis AF, Igoumenou VG, Megaloikonomos PD, Panagopoulos GN, Papagelopoulos PJ, et al. (2017) Giant cell tumour of bone revisited. SICOT J 3: 54. [Crossref]
- Çomunoğlu N, Kepil N, Dervişoğlu S (2019) Histopathology of giant cell tumour of bone: with special emphasis on fibrohistiocytic and aneurysmal bone cyst like components. Acta Orthop Traumatol Turc 53: 35-39. [Crossref]
- Chan CM, Adler Z, Reith JD, Gibbs CP Jr (2015) Risk factors for pulmonary metastases from giant cell tumour of bone. J Bone Joint Surg Am 97: 420-428. [Crossref]
- Presneau N, Baumhoer D, Behjati S, Pillay N, Tarpey P, et al. (2015) Diagnostic value of H3F3A mutations in giant cell tumour of bone compared to osteoclast- rich mimics. J Pathol Clin Res 1: 113-123. [Crossref]
- Lüke J, von Baer A, Schreiber J, Lübbehüsen C, Breinin T, et al. (2017) H3F3A mutation in a giant cell tumour of bone is detected by immune histochemistry using a monoclonal antibody against G34W mutated site of the histone H3.3 variant. Histopathology 71: 125-133. [Crossref]
- Yang Y, Huang Z, Niu X, Xu H, Li Y, et al. (2017) Clinical characteristics and risk factor analysis of lung metastasis of benign giant cell tumour of bone. J Bone Oncol 7: 23-28. [Crossref]
- Image 1 Courtesy: Medbullets.com
- Image 2 Courtesy: Wikipedia 1
- Image 3 Courtesy: aiaohns.in
- Image 4 Courtesy: Research gate
- Image 5 Courtesy: Orthopaedics one.com
- Image 6 Courtesy: Orthobullets.com
- Image 7 Courtesy: Pathology outlines
- Image 8 Courtesy: I Love Pathology.com