Current knowledge on Coronavirus disease 2019 (COVID-19) pathogenesis leads to speculate on the occurrence of antibody dependent enhancement (ADE) in severe acute respiratory syndrome coronavirus (SARS-CoV)-2 infection with mechanisms inferred by those postulated for SARS-CoV: in patients with previous exposure to endemic coronaviruses, a robust T helper (Th)1-dependent neutralizing antibody response, elicited before viral clearance, may lead to a change in virus cell tropism. Direct infection of monocytes/macrophages and B cells could determinate an immune enhancement of the disease, characterized by the skew of macrophage response and subsequent over-secretion of interleukin (IL)-6, IL-8 and programmed death 1 ligand, leading to T cells reduction and exhaustion and M1 cells accumulation in lungs, responsible for severe and persistent lung injury. The occurrence of ADE should be taken into account not only to assess the safety of candidate vaccines but also to assess safety and efficacy of investigational immunomodulatory treatments: if ADE occurs, hydroxy(chloroquine) could worsen severity of COVID-19 cases by enhancing SARS-CoV-2 internalization in B cells and inhibiting M2 polarization of macrophages in lungs. On the contrary, ulinastatin could effectively switch back the skew of macrophage response, while thymosin alpha 1 (Tα1) could revert T cell reduction and exhaustion, and prime Th2 cell differentiation. Dysregulation of type-1/type-2 immune response may be a pathogenic mechanism underlying the occurrence of ADE in several viral infections, such as dengue hemorrhagic fever, ebola virus infection and other viral hemorrhagic fevers. Therefore, co-administration of Tα1 and ulinastatin could be investigated not only in treating severe COVID-19 patients but also for other deadly viral diseases for which ADE of infection has been demonstrated or postulated.
antibody dependent enhancement (ADE), COVID-19, hydroxychloroquine, SARS-CoV-2, thymosin alpha 1, type-1/type-2 immune response, ulinastati
Coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus (SARS- CoV)-2 infection, presents a broad clinical spectrum, ranging from asymptomatic-mild to fatal cases [1]. Disease severity is attributable to an abnormal host response [2] resembling the pathophysiology of SARS- CoV infection [3]. As previously hypothesized for SARS patients, the occurrence of antibody dependent enhancement (ADE) in SARS-CoV-2 infection has been claimed as a possible explanation for differences in clinical presentation and in geographic fatality rate [4,5].
ADE of infection is exploited by a wide range of viruses and is thought to occur when the virus is bound by non-neutralizing antibodies or sub-neutralizing concentrations of antibodies which, instead of neutralizing it, facilitate virus internalization into host cells (extrinsic ADE) [6]. Internalized antibody-virus immunocomplexes may modulate the innate antiviral response to increase virus production (intrinsic ADE) [6]. The ADE phenomenon is thought to prompt massive release of inflammatory and vasoactive mediators that ultimately contribute to disease severity [6]. Extrinsic ADE is mainly mediated by crystallizable fragment-gamma receptor (FcγR)-expressing cells that bind the Fc portion of circulating immunoglobulin (Ig)G antibodies produced during primary infection, therefore it requires a previous sensitization of the humoral immune response to specific viral epitopes [6]. However, in West Nile virus infection, an IgG- independent ADE mechanism was described for the first time: the enhancement of the infection was related to the interaction between complement receptors and IgM antibodies; the two enhancement pathways, IgG and IgM-related, were independent but were not mutually exclusive and could act synergistically [7]. Also in Ebola virus infection, both pathways have been described in vitro: it has been shown that IgM level was correlated with complement component 1q (C1q)/C1q receptors-dependent ADE, while IgG2a level with the FcγR-dependent mechanism. Interestingly, while IgG2a antibodies triggered ADE, IgG1 antibodies exerted a neutralizing effect: therefore, only the response towards the T helper (Th)1 phenotype was associated with ADE [8].
While for many viruses, ADE increases the viral load by infecting a higher number of already susceptible cells [9], ADE of SARS-CoV infection is characterized by a change in cell tropism: anti-Spike protein IgG antibodies (S-IgG) neutralize virus entry into the angiotensin converting enzyme 2 (ACE2)-bearing cells (target cells), and enhance virus internalization into macrophages, monocytes and B cells through an IgG/FcγRII–dependent, ACE2-indipendent mechanism [10-14] (Figure 1). Not all the FcγRII-bearing cells are permissive to ADE of SARS-CoV infection which seems to depend on the intracellular FcγRII domain and downstream signaling rather than on extracellular binding affinity or immune-complexes internalization rate [14]. Infection of immune cells appears to be self-limiting or abortive [10-13], however, given the ability of intracellular innate immune sensors to detect viral gene species and the disturbance of cellular homeostasis caused by SARS-CoV proteins, a central role of B cells has been postulated in the pathogenic mechanism of ADE [11, 12]. Although preliminary studies have found no changes in gene expression of pro- inflammatory cytokines/chemokines of infected macrophages [13], a recent research has clarified the role of macrophages polarization in ADE of SARS-CoV pathogenesis [15]. In response to various signals, macrophages may undergo classical M1 activation -stimulated by Toll-like receptor (TLR) ligands and interferon-gamma (IFN-γ)- or alternative M2 activation -stimulated by interleukin (IL)-4/IL-13- mirroring the Th1–Th2 polarization of T cells [16]. The phenotype of polarized M1-M2 macrophages can be reversed: classically activated M1 cells are involved in initiating and sustaining inflammation while M2 or M2-like cells are associated with the resolution process [17]. Liu and colleagues, using a SARS-CoV-macaque model, showed a significantly enhanced and persistent lung damage, despite viral suppression, in the S–IgG pretreated group compared with the control group: in macaques not treated with S-IgG, alveolar monocytes/macrophages assumed a M2 function as early as 2 days post infection leading to the resolution of lung damage and restoration of homeostasis, on the contrary S–IgG treatment skewed the macrophage response in the lungs leading to persistence of pro-inflammatory M1 cells, uncontrolled inflammation and tissue injury [15]. The authors, conducting a temporal analysis of productive viral infection, antibody response, and macrophage functional changes during the first week of infection, observed that the skew towards the M1 cells was present only when the neutralizing antibody (NAb) response had been elicited before the virus clearance [15]. Histopathological examination of deceased SARS patients showed findings similar to those found in ADE-SARS-CoV macaques, with pro-inflammatory M1 lung infiltration and absence of M2 cells [15]. S-IgG sera had no effect on M1 cells of deceased patients, but caused a dose-dependent increase in the production of IL-8, IL-6 and monocyte chemo-attractant protein 1 (MCP1) in M2 cells [15]. Similar results were observed when treating M2 cells of SARS patients, obtained during the acute phase of the infection, with antisera from deceased SARS patients: IL-8 production was correlates with the NAb titers of sera [15]. Interestingly, all but one antisera from recovered SARS patients did not elicit cytokine overproduction in M2 cells, the only M2 skewing sera had higher NAb titer (similar to those found in deceased patients) compared to the others [15]. These latter findings are consistent with the observation that an early NAb response (and a concomitant robust Th1 response) is a marker of poor disease outcome in SARS patients [18,19], while clinically mild cases have relatively low antibody titers and a less sustained immune response [20,21]. Taking into account serological cross-reaction between SARS-CoV and human endemic coronaviruses (HCoVs), the priming effect of a previous HCoV infection could be responsible for an early IgG response in severe cases of SARS [18]. In conclusion, the occurrence of ADE in SARS-CoV infection seems to depend on a previous exposure to HCoVs, responsible for an early sero-conversion, and on a concomitant robust Th1 response. The phenomenon could lead to a change in SARS-CoV cell tropism with direct infection of monocytes/macrophages and B cells, through an ACE2-indipendent mechanism, and consequently to an immune enhancement of the disease which causes the inhibition of the M2 cells activation and the accumulation of M1 cells in the lungs, responsible for severe and persistent lung injury.
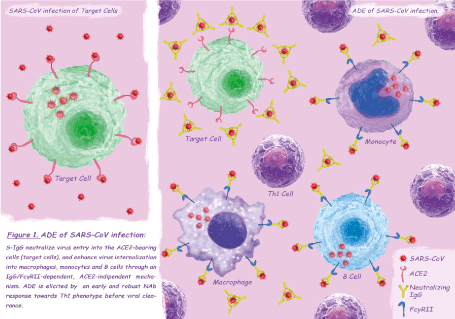
Figure 1. ADE of SARS-CoV infection
ADE of SARS-CoV2 infection
The pathophysiology of SARS-CoV-2 infection seems to be closely related to that of SARS-CoV infection: hystopathological examination of deceased patients showed macrophage accumulation in alveolar spaces and fibroproliferative areas in the lungs, and also the staging of the disease resembles that of SARS with exudative diffuse alveolar damage (DAD) in the early stage and organizing and fibrotic DAD in the late phase [2,22,23]. Severe cases are characterized by an exuberant release of pro-inflammatory cytokines and lymphocytopenia, in particular, serum increase of IL-6 and IL-8 is associated with the progression of the disease [2] and the extension of T lymphocytopenia [22]. Lymphocytopenia is directly related to the expression of programmed cell death protein-1 (PD-1) on T cells surface, a marker of T cell exhaustion [24,25]. Interestingly, the correlation between T cell exhaustion and disease severity has also been demonstrated in Ebola virus infection [26]. Alveolar macrophages of COVID-19 deceased patients exhibit a broad and strong expression of PD-1 ligand (PD-L1) [22], therefore pulmonary inflammation could trigger the PD-1/PD-L1 axis leading to T cell exhaustion. The strong PD-L1 up-regulation represents an indirect marker of the classic M1 activation [27], hence, similarly to the ADE-SARS-macaque model, also in SARS- CoV-2 infection the inhibition of the M2 response and the accumulation of M1 cells could be responsible for severe lung injury (Figure 2). As previously described for SARS-CoV infection, in COVID-19 patients macrophages may be directly affected by the virus [2,22], and it seems that B cells play a crucial role in the pathogenesis of severe cases [28].
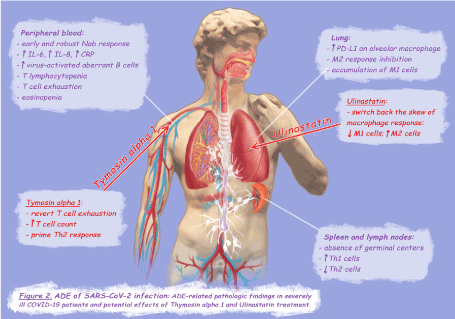
Figure 2. ADE of SARS-CoV-2 infection
An interesting study on the adaptive immune response to SARS-Cov-2 infection in severe COVID-19 patients showed, by analyzing lympho nodes and spleens of deceased patients, the absence of germinal centers, a substantial increase in Th1 cells and a consistent reduction in Th2 cells [29]. In addition, an extra-germinal center/extra-follicular type class-switched antibody response to SARS-CoV-2 was detected, whit significant accumulation of virus-activated aberrant B cells in tissues and blood of severely ill patients -included switched memory B cells- [29] (Figure 2). Sera from patients with a recent history of HCoVs infection have been found to have SARS-CoV-2 neutralization activity [30], which could explain why in some COVID-19 patients has been observed an earlier appearance of IgG compared to IgM, indicating a secondary immune response to a cross-reactive antigen [31]. Therefore, the reason that older patients recovered from SARS- Cov-2 infection have significantly higher NAb titers than younger patients [32], could be that they have been exposed to previous HCoVs infections more times with respect to young subjects. In patients recovered from mild COVID-19, NAb titers at discharge were positively correlated with blood CRP levels but negatively correlated with lymphocyte counts at admission, suggesting that high levels of NAbs could be related to a strong inflammation [32]. This hypothesis is corroborated by the evidence that in patients admitted to Intensive Care Unit (ICU) the NAb response rose significantly earlier and to a much greater extent than in mild cases [33].
Previous exposure to cross-reactive antigen seems to be a necessary but not sufficient condition for the occurrence of ADE in both SARS-CoV and Ebola virus infection: in fact, only a strong antibody response towards the Th1 phenotype appears to be associated with the enhancement of the disease [8,18,19]. Of note, obesity, type 2 diabetes, and hypertension, which represent risk factors for severe COVID-19 [34,35], are all clinical conditions characterized by chronic type 1 polarization of immune cells [36-38], on the contrary, history of allergy, a Th2-biased immune response condition, seems to protect from clinically severe infection [39,40]. Furthermore, eosinopenia is considered a marker of poor clinical outcome [41] while an increase in serum eosinophils has been suggested as an indicator of clinical improvement [42]. All the above mentioned evidence leads to speculate on the occurrence of ADE in SARS-CoV-2 infection with mechanisms inferred by those postulated for SARS-CoV: in COVID-19 patients with previous exposure to HCoVs, an early and robust NAb response towards the Th1 phenotype, elicited before viral clearance, may lead to a change in virus cell tropism. Direct infection of monocytes/macrophages and B cells could determinate an immune enhancement of the disease, characterized by the inhibition of M2 response and subsequent over-secretion of IL-6, IL-8 and PD-L1, leading to reduction and exhaustion of T cells, and M1 cells accumulation and persistence in lungs (Figure 2).
Hydroxychloroquine
The occurrence of ADE in SARS-CoV-2 infection should be taken into account not only to assess the safety of candidate vaccines [43] but also to evaluate the safety and efficacy of investigational immunomodulatory treatments. Among others, the 4-aminoquinolines chloroquine (CQ) and hydroxychloroquine (HCQ) have been widely used for their alleged antiviral and anti-inflammatory effects.
Preliminary results from a multi-centre, randomized, controlled trial showed that in hospitalized COVID-19 patients, hydroxychloroquine use was associated with a longer length of hospital stay and an increased risk of progressing to invasive mechanical ventilation or death [44]. Particular attention was paid to drug- induced QTc interval prolongation and ventricular arrhythmias, which seem to be higher than expected in COVID-19 patients treated with 4-aminoquinolines [45]; however, beyond drug-induced cardiotoxicity, it has been hypothesized that they could worsen COVID-19 severity through other mechanisms [46,47]. The antiviral efficacy of CQ has been inferred by in vitro experiments on SARS-CoV infection: it has been shown that increasing endosomal pH and altering ACE2 glycosylation prevent viral entry [48], but while the increased endosomial pH inhibits viral entry into target cells, it enhances SARS-CoV internalization in B cells, in an ACE2-independent mechanism [12]. Regarding the immunomodulatory effects of the antimalarial compounds, CQ inhibits M2 polarization in two ways: first, it inhibits T-cell proliferation by reducing IL-2 production and IL-2 responsiveness, which play a crucial role in “priming” T cells for Th2 cell differentiation [47]; secondly, by neutralizing lysosomal ph, it inhibits lysosomal lipolysis in macrophages, a fundamental process in IL-4-dependent M2 activation pathway [49]. Therefore, if ADE occurs in severe COVID-19 patients, CQ and HCQ could worsen the clinical course by further enhancing the same pathogenic mechanisms responsible for ADE. The high percentage of QTc interval prolongation and ventricular arrhythmias may not merely be a direct drug-side effect of the 4-aminoquinoline, rather it could represent a sign of myocardial injury secondary to an immune enhancement of the disease induced by the treatment. Interestingly, no cardiac side effects have been reported among mild COVID-19 cases treated with HCQ [50], while imbalance in Th1/Th2 response has already been proposed as a mechanism of myocardial injury in severe SARS-CoV-2 infection [51]. In conclusion, while the antimalarial administration, although inefficient, may not have harmful consequences on mild cases [50], it seems to exert detrimental effect on severe COVID-19 patients [44], for whom ADE is likely to occur.
Thymosin alpha 1
A small observational study has shown that thymosin alpha 1 (Tα1) administration significantly reduces the mortality of severe COVID-19 patients by restoring T cell count and reducing PD-1 expression on T cells surface [25]. Although the thymic peptide Tα1 exhibits pleiotropic effects on the immune system, T-cell proliferation and prevention of T-cell exhaustion seems to be related to an increase in IL-2 production and IL-2 responsiveness [52]: opposite effect that CQ exerts on T cells. Interestingly, Tα1 treatment in sepsis patients has shown a beneficial effect on mortality, restoring peripheral T cell counts and balancing pro- inflammatory and anti-inflammatory cytokines, with a reduction in serum tumor necrosis factor-alpha (TNF- α) level and an increase in IL-10 [53]. Also in sepsis, in fact, an unbalanced pro-inflammatory response seems to be responsible for tissue/organ damage and, similarly to what observed in COVID-19 patients, history of allergy seems to be a protective factor [54]. Krishack and colleagues evaluated Th cell polarization among patients with S. aureus bacteremia, showing that survivors had a higher percentage of circulating Th2 cells early in the course of infection [54]. The authors, using an animal model, elucidated the protective benefit of type 2 immune response and subsequent eosinophilia in counterbalancing the pathogen-induced pro-inflammatory response [54]. Other interesting findings from the cited study were that sepsis-related mortality may result from the inflammatory response specifically in the lungs, and that the driver of inflammation seems to be intrinsic to the host response itself, rather than relate to the persistence of bacteria [54].
Ulinastatin
The immune rebalancing effect of Tα1 on septic patients appears to be potentiated when co-administered with ulinastatin [55,56]. Ulinastatin is a urinary trypsin inhibitor obtained by separation and purification from male urine, which inhibits the production of inflammatory factors and the release of inflammatory mediators, and improves the imbalance of the inflammatory microenvironment [57]. Zhang et colleagues, in a small randomized clinical trial (RCT) on sepsis arising from intra-abdominal infection due to carbapenem-resistant bacteria, have shown that co-administration of Tα1 and ulinistatin improves survival rate and is associated with a shorter duration of mechanical ventilation, a shorter duration of dopamine therapy, and a shorter ICU stay: on day 8, the size of the T cell population was greater and IL-4 and IL-10 levels were higher in the treated group compared to the control group, while the levels of TNF-α, IL-1, and IL-6 were lower [56]. Liu and colleagues, by using an experimental mouse model, investigated the protective effect of ulinastatin on severe pulmonary infection under immunosuppression: the model group presented severe lung injury with alveolar edema and macrophage infiltration, while most of the lung tissue in the ulinastatin group remained intact [57]. Bronchoalveolar lavage fluid in the model group showed a significant increase in IL-6, TNF-α and IL-1β, and a significant decrease in IL-4, IL-10, and IL-13, while in the treated group the levels of pro-inflammatory and anti-inflammatory cytokines returned to normal level [57]. Finally, by comparing macrophage polarization in lungs, the model group showed a significant higher concentration of M1 cells and a significant lower amount of M2 cells with respect to ulinistatin group: the authors concluded that ulinastatin may exert a protective effect on lung injury of immunosupressed mice by regulating the inflammatory microenvironment through polarization from M1 into M2 macrophages [57]. Due to its immunomodulatory properties, the urinary trypsin inhibitor has been proposed as a potential candidate for COVID-19 treatment [58] but studies on its efficacy has not yet been carried out.
In conclusion, ulinastatin could effectively switch back the skew of macrophage response that could occurs in ADE-SARS-CoV-2 patients, while Tα1 could restore T cell count, revert T cell exhaustion, and prime Th2 cell differentiation (Figure 2).
The immune imbalance in ADE of other viral infections
Intriguingly, the imbalance between pro-inflammatory cytokines IL-6 and IL-8 and the anti-inflammatory cytokine IL-10 seems to be related to the hemorrhagic manifestations of dengue patients [59]; therefore, dysregulation of type-1/type-2 immune response could be a pathogenic mechanism underlying the occurrence of ADE in several viral infections. Hence, taking into account the lack of specific treatments and vaccines, co-administration of Tα1 and ulinastatin could be investigated not only for highly pathogenetic human coronaviruses but also for other deadly viral diseases for which ADE of infection has been demonstrated or postulated, such us dengue hemorrhagic fever, ebola virus infection and other viral hemorrhagic fevers.
In conclusion, further studies addressing the occurrence of ADE in SARS-CoV-2 infection are needed, not only to evaluate safety of candidate vaccines, but also for searching effective immunomodulatory treatments. Due to the known safety profile of the two compounds [55,56,58] and the potential occurrence of ADE of infection in COVID-19 cases, large RCTs should be conducted to evaluate the efficacy of Tα1 and ulinastatin in treating severe COVID-19 patients. The effectiveness of the two drugs could also be investigated in other deadly viral diseases for which ADE of infection has been demonstrated or postulated.
No funding or sponsorship was received for this study or publication of this article.
Maurizio Guastalegname, Laura D’argenio and Alfredo Vallone meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Maurizio Guastalegname, Laura D’argenio and Alfredo Vallone declare that they have no conflict of interest.
- Zhang H, Shang W, Liu Q, Zhang X, Zheng M, et al. (2020) Clinical characteristics of 194 cases of COVID-19 in Huanggang and Taian, China. Infection 1-8. [Crossref]
- Li S, Jiang L, Li X (2020) Clinical and pathological investigation of patients with severe COVID-19. JCI Insight 5: 138070. [Crossref]
- Petrosillo N, Viceconte G, Ergonul O, Ippolito G, Petersen E, et al. (2020) COVID-19, SARS and MERS: are they closely related? Clin Microbiol Infect 26: 729-734. [Crossref]
- Tetro JA (2020) Is COVID-19 receiving ADE from other coronaviruses?. Microbes Infect 22: 72-73. [Crossref]
- Cegolon L, Pichierri J, Mastrangelo G (2020) Hypothesis to explain the severe form of COVID-19 in Northern Italy. BMJ Glob Health 5: e002564.
- Taylor A, Foo SS, Bruzzone R, Dinh LV, King NJ, et al. (2015) Fc receptors in antibody-dependent enhancement of viral infections. Immunol Rev 268: 340-364.
- Cardosa MJ, Porterfield JS, Gordon S (1983) Complement receptor mediates enhanced flavivirus replication in macrophages. J Exp Med 158: 258-263.
- Takada A, Ebihara H, Feldmann H, Geisbert TW, Kawaoka Y, et al. (2007) Epitopes required for antibody-dependent enhancement of Ebola virus infection. J Infect Dis 196 Suppl 2: S347-S356. [Crossref]
- Sullivan NJ (2001) Antibody-mediated enhancement of viral disease. Curr Top Microbiol Immunol 260: 145-169.
- Li L, Wo J, Shao J (2003) SARS-coronavirus replicates in mononuclear cells of peripheral blood (PBMCs) from SARS patients. J Clin Virol 28: 239-244. [Crossref]
- Kam YW, Kien F, Roberts A (2007) Antibodies against trimeric S glycoprotein protect hamsters against SARS-CoV challenge despite their capacity to mediate FcgammaRII-dependent entry into B cells in vitro. Vaccine 25: 729- 740.
- Jaume M, Yip MS, Cheung CY (2011) Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH- and cysteine protease-independent FcγR pathway. J Virol 85: 10582-10597.
- Yip MS, Leung HL, Li PH (2016) Antibody-dependent enhancement of SARS coronavirus infection and its role in the pathogenesis of SARS. Hong Kong Med J 22: 25-31. [Crossref]
- Yip MS, Leung NH, Cheung CY (2014) Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol J 11: 82.
- Liu L, Wei Q, Lin Q (2019) Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 4: e123158.
- Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122: 787-795.
- Martinez FO, Helming L, Gordon S (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27: 451-483.
- Ho MS, Chen WJ, Chen HY (2005) Neutralizing antibody response and SARS severity. Emerg Infect Dis 11: 1730-1737.
- Lee N, Chan PK, Ip M (2006) Anti-SARS-CoV IgG response in relation to disease severity of severe acute respiratory syndrome. J Clin Virol 35: 179-184.
- Tso EY, Tsang OT, Lam B, Ng TK, Lim W, et al. (2004) Natural course of severe acute respiratory syndrome-associated coronavirus immunoglobulin after infection. J Infect Dis 190: 1706-1707.
- Wilder-Smith A, Teleman MD, Heng BH, Earnest A, Ling AE, et al. (2005) Asymptomatic SARS coronavirus infection among healthcare workers, Singapore. Emerg Infect Dis 11: 1142-1145.
- Wang C, Xie J, Zhao L (2020) Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine 57: 102833.
- Nunes Duarte-Neto A, de Almeida Monteiro RA, Da Silva LFF (2020) Pulmonary and systemic involvement of COVID-19 assessed by ultrasound-guided minimally invasive autopsy. Histopathology 77: 186-197. [Crossref]
- Diao B, Wang C, Tan Y (2020) Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front Immunol 11: 827. [Crossref]
- Liu Y, Pang Y, Hu Z (2020) Thymosin alpha 1 (Tα1) reduces the mortality of severe COVID-19 by restoration of lymphocytopenia and reversion of exhausted T cells. Clin Infect Dis 71: 2150-2157. [Crossref]
- Ruibal P, Oestereich L, Lüdtke A (2016) Unique human immune signature of Ebola virus disease in Guinea. Nature 533: 100-104. [Crossref]
- Loke P, Allison JP (2003) PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A 100: 5336-5341. [Crossref]
- Quinti I, Lougaris V, Milito C (2020) A possible role for B cells in COVID-19? Lesson from patients with agammaglobulinemia. J Allergy Clin Immunol 146: 211-213.e4. [Crossref]
- Kaneko N, Kuo HH, Boucau J (2020) Massachusetts Consortium on Pathogen Readiness Specimen Working Group. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 183: 143-157.e13.
- Van der Heide V (2020) SARS-CoV-2 cross-reactivity in healthy donors. Nat Rev Immunol 20: 408. [Crossref]
- Fierz W, Walz B (2020) Antibody Dependent Enhancement Due to Original Antigenic Sin and the Development of SARS. Front Immunol 11: 1120. [Crossref]
- Wu F, Liu M, Wang A (2020) Evaluating the Association of Clinical Characteristics with Neutralizing AntibodyLevels in Patients Who Have Recovered from Mild COVID-19 in Shanghai, China. JAMA Intern Med 180: 1356-1362. [Crossref]
- Liu L, To KK, Chan KH (2020) High neutralizing antibody titer in intensive care unit patients with COVID-19. Emerg Microbes Infect 8: 1-30.
- Denova-Gutiérrez E, Lopez-Gatell H, Alomia-Zegarra JL (2020) The association between obesity, type 2 diabetes, and hypertension with severe COVID-19 on admission among Mexicans. Obesity (Silver Spring). [Crossref]
- Chen Q, Zheng Z, Zhang C (2020) Clinical characteristics of 145 patients with corona virus disease 2019 (COVID-19) in Taizhou, Zhejiang, China. Infection 48: 543-551. [Crossref]
- Wu H, Ballantyne CM. Metabolic Inflammation and Insulin Resistance in Obesity. Circ Res 126: 1549- 1564.
- Zhou T, Hu Z, Yang S, Sun L, Yu Z, et al. (2018) Role of Adaptive and Innate Immunity in Type 2 Diabetes Mellitus. J Diabetes Res 2018: 7457269.
- Idris-Khodja N, Mian MO, Paradis P, Schiffrin EL (2014) Dual opposing roles of adaptive immunity in hypertension. EurHeart J 35: 1238-1244. [Crossref]
- Shi W, Gao Z, Ding Y, Zhu T, Zhang W, et al. (2020) Clinical characteristics of COVID-19 patients combined with allergy. Allergy 75: 2405-2408. [Crossref]
- Liu S, Zhi Y, Ying S (2020) COVID-19 and Asthma: Reflection During the Pandemic. Clin Rev Allergy Immunol 59: 78-88 [Crossref]
- Qin C, Zhou L, Hu Z (2020) Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis 71: 762-768. [Crossref]
- Liu F, Xu A, Zhang Y (2020) Patients of COVID-19 may benefit from sustained Lopinavir-combined regimen and the increase of Eosinophil may predict the outcome of COVID-19 progression. Int J Infect Dis 95: 183-191.
- Arvin AM, Fink K, Schmid MA (2020) A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature. [Crossref]
- Horby P, Mafham M, Linsell L (2020) Effect of Hydroxychloroquine in Hospitalized Patients with COVID-19: Preliminary results from a multi-centre, randomized, controlled trial. medRxiv 383: 2030-2040
- Jankelson L, Karam G, Becker ML, Chinitz LA, Tsai MC, et al. (2020) QT prolongation, torsades de pointes, and sudden death with short courses of chloroquine or hydroxychloroquine as used in COVID-19: A systematic review. Heart Rhythm S1547-5271: 30431-30438.
- Touret F, de Lamballerie X (2020) Of chloroquine and COVID-19. Antiviral Res 177: 104762.
- Guastalegname M, Vallone A (2020) Could chloroquine /hydroxychloroquine be harmful in Coronavirus Disease 2019 (COVID-19) treatment?. Clin Infect Dis 71: 888-889. [Crossref]
- Vincent MJ, Bergeron E, Benjannet S (2005) Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J 2: 69.
- Huang SC, Everts B, Ivanova Y (2014) Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15: 846-855.
- Mitjà O, Corbacho-Monné M, Ubals M (2020) Hydroxychloroquine for Early Treatment of Adults with Mild Covid-19: A Randomized-Controlled Trial. Clin Infect Dis. ciaa1009. [Crossref]
- Zheng YY, Ma YT, Zhang JY, Xie X (2020) COVID-19 and the cardiovascular system. Nat Rev Cardiol 17: 259-260.
- Baxevanis CN, Frillingos S, Seferiadis K (1990) Enhancement of human T lymphocyte function by prothymosin alpha: increased production of interleukin-2 and expression of interleukin-2 receptors in normal human peripheral blood T lymphocytes. Immunopharmacol Immunotoxicol 12: 595-617.
- Liu F, Wang HM, Wang T, Zhang YM, Zhu X, et al. (2016) The efficacy of thymosin α1 as immunomodulatory treatment for sepsis: a systematic review of randomized controlled trials. BMC Infect Dis 16: 488.
- Krishack PA, Louviere TJ, Decker TS (2019) Protection against Staphylococcus aureus bacteremia-induced mortality depends on ILC2s and eosinophils. JCI Insight 4: e124168. [Crossref]
- Chen H, He MY, Li YM (2009) Treatment of patients with severe sepsis using ulinastatin and thymosin alpha1: a prospective, randomized, controlled pilot study. Chin Med J (Engl) 122: 883-888. [Crossref]
- Zhang Y, Chen H, Li YM (2008) Thymosin alpha1- and ulinastatin-based immunomodulatory strategy for sepsis arising from intra-abdominal infection due to carbapenem-resistant bacteria. J Infect Dis 198: 723-730. [Crossref]
- Liu W, Pang G, Wang S, Sun A (2017) Protective effect of ulinastatin on severe pulmonary infection under immunosuppression and its molecular mechanism. Exp Ther Med 14: 3583-3588.
- Ye Q, Wang B, Mao J (2020) The pathogenesis and treatment of the `Cytokine Storm' in COVID-19. J Infect 80: 607-613.
- Iani FC, Caldas S, Duarte MM (2016) Dengue patients with early hemorrhagic manifestations lose coordinate expression of the anti-inflammatory cytokine il-10 with the inflammatory cytokines IL-6 and IL-8. Am J Trop Med Hyg 95: 193-200. [Crossref]